
More Information
Submitted: April 21, 2026 | Accepted: May 14, 2026 | Published: May 15, 2026
Citation: Puccio G, et al. Proposal for a Study Protocol on the Use of Antioxidants as Adjuvants in Cancer Therapy. Arch Case Rep. 2026; 10(5): 43-57. Available from:
https://dx.doi.org/10.29328/journal.acr.1001188
DOI: 10.29328/journal.acr.1001188
Copyright license: © 2026 Puccio G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Phosphoethanolamine; Phosphatidylethanolamine; ETNK-1; NRF2; mTOR; AMPK; FSP-1; GSH; CYP450; Csncerogenesis

Proposal for a Study Protocol on the Use of Antioxidants as Adjuvants in Cancer Therapy
Giovanni Puccio1 and Massimo Coppolino2*
1Researcher and President of the Emmanuele Free Research Association, Palermo, Italy
2Molecular Biologist, Scientific Director of the Emmanuele Free Research Association and Associate Professor for Mycrobiology and Genetics at IMA University, Italy
*Corresponding author: Dott. Massimo Coppolino, Adjunct Professor, Immunology, Molecular Biologist, MSc in Biochemistry and Clinical Nutrition, IMA University, Italy, Email: [email protected]
A protocol is proposed for studying the effects of oxidant substances, such as GSH, NAC, and vitamin C, in combating tumors as an adjuvant to chemotherapy. These same substances are combined, at different times and in different cases, with molecules such as phosphatidylethanolamine and ivermectin, and the use of alternative techniques such as oxigen-ozone Therapy o byorisonance. These molecules are currently being studied for their efficacy at various research centers. Given their biochemical compatibility, the author has decided to combine these with antioxidants and their proven activity as catalysts of antitumor processes.
The protocol draws not only on the abundant literature confirming that the use of antioxidants, with precise timing of administration, can facilitate recovery from cancer when combined with chemotherapy, but also that they can drastically reduce both the duration of treatment and hospitalization and death rates. Furthermore, the author, who has analyzed numerous cases, has noted that it leads to an almost complete reduction in side effects, lowering chemotherapy-induced toxicosis. Relapses also appear to be reduced compared to the average.
For this reason, we will first analyze the individual substances and how they interact with each other, bridging different cellular life cycles that only apparently appear unrelated but which, as has been demonstrated, are the basis for the development of many forms of cancer.
Phosphoethanolamine and phosphatidylethanola-mine: Role in carcinogenesis the main difference between PE and PEA concerns chemical structure and complexity:
- Phosphoethanolamine (PEA) is a simple precursor, composed of a phosphate group linked to ethanolamine. It is hydrophilic (water-soluble).
- Phosphatidylethanolamine (PE) is a complete phospholipid. It is formed when phosphoethanolamine binds to a diacylglycerol molecule. It is a fundamental component of cell membranes and is amphipathic (has a soluble "head" and fatty "tails").
In short: the former is a “building block,” the latter is the finished “wall” of the cell.
Free radicals damage these molecules through lipid peroxidation, but with different effects:
- Phosphatidylethanolamine (PE), being a structural component of membranes rich in polyunsaturated fatty acids (PUFA), it is the primary target. Free radicals attack the fatty "tails," triggering a chain reaction that stiffens and destroys the cell membrane.
- Phosphoethanolamine, being a water-soluble precursor with no fatty chains, it does not directly undergo peroxidation. However, oxidative stress can alter the enzymes that convert it to PE, reducing the cell's ability to repair its membranes.
In summary: radicals destroy membranes (PE) and block repair processes (phosphoethanolamine). Oxidative stress and phospholipid degradation contribute to carcinogenesis through three main mechanisms:
1. Membrane instability and signalingPhosphatidylethanolamine (PE) peroxidation alters membrane fluidity. This distorts surface receptors, sending incorrect growth signals to the nucleus that stimulate uncontrolled cell proliferation.
2. Formation of toxic aldehydes
The breakdown of oxidized fats generates byproducts such as malondialdehyde (MDA). These molecules migrate from the membrane to DNA, binding to it and causing direct genetic mutations in tumor suppressor genes.
3. Evasion of apoptosis
Altering the ratio of phosphoethanolamine to PE can block autophagy and apoptosis (programmed cell death). Damaged cells, which should die, continue to live and replicate, accumulating further genomic damage. This part of the cancer cycle is that one who needs more concentraton of antioxidant, because the increas of mutations do not signifize increasing of cancer growth.
It is now established that mutations during carcinogenesis are not random, but are guided towards specific biochemical pathways by changes in the structure of the cell membrane, the true “brain” and “decision-making center” of the cell, acting as a regulator of gene expression in response to chemical and energetic balances between the intercellular and extracellular environments. The cell is not at the service of the organism; each cell is an entity in its own right, as were their progenitor protozoa, capable of surviving in isolation. Therefore, if it senses that the environment around it is unfavorable to its survival, it simply activates genes selected through the activation of particular intramembrane receptors that expose them inward. These receptors provide either the synthesis of necessary corrective enzymes (proteinases, RNAses, etc.) or by stimulating the synthesis of genes that reactivate mitosis, generating the tumor.
Phosphatidylethanolamine (PE) is essential for the initiation of autophagy, the process by which the cell “cleans up” its damaged components. Specifically, the LC3 protein must chemically bind to PE (lipidation process) to form the autophagosome, the vesicle that engulfs cellular waste.
In cancer cells, this mechanism is often manipulated:
- Initial suppression: the tumor reduces autophagy to prevent oxidative stress from killing precancerous cells.
- Advanced survival: once stabilized, the tumor can increase PE turnover to recycle nutrients and resist chemotherapy or hypoxia. In this period a low-fat diet in necessary to block the process.
- Cell signaling: altered PE levels affect also the stability of mitochondrial membranes, regulating the release of signals that induce apoptosis (cell death).
If PE is damaged by free radicals, autophagy becomes inefficient, allowing the accumulation of defective organelles that fuel inflammation and cancer progression.
Cancer drugs that target the bond between LC3 and phosphatidylethanolamine (PE) act in two main ways to disrupt the tumor cell:
- Autophagy inhibitors: Some compounds block the enzyme (ATG7) responsible for the binding of LC3 to PE. Without this "anchor," the tumor cannot recycle nutrients and succumbs to metabolic stress.
- Ferroptosis inducers: New therapies aim to selectively oxidize PE-rich membranes. This triggers cell death due to excess free radicals, to which cancer cells are particularly sensitive.
In practice, the aim is to sabotage garbage trucks (autophagosomes) or explode the tumor’s storage facilities (membranes).
The drugs target metabolic selectivity by exploiting the differences between healthy and tumor cells:
- Warburg effect and hyperactivation: cancer cells have an accelerated metabolism and produce significantly more free radicals. Drugs (such as ferroptosis inducers) act like a "straw that breaks the camel's back": healthy cells have intact antioxidant defenses to manage stress, while cancer cells are already stretched to their limits and collapse.
- Autophagy dependence: a normal cell can temporarily survive without recycling its components. A cancer cell, which grows rapidly in nutrient-poor environments, is "dependent" on the LC3-PE linkage for energy; blocking this link starves the tumor but leaves healthy cells in a state of safe quiescence.
- Specific targeting: some new delivery systems (nanoparticles) release the active ingredient only in the presence of an acidic or hypoxic environment, characteristics typical of tumor mass and absent in healthy tissue.
Healthy cells survive these treatments (and daily oxidative stress) thanks to a genetic arsenal that cancer cells have often mutated or “exacerbated” to the point of failure.
The main “shield” genes are:
1. GPX4 (Glutathione Peroxidase 4)
It is the supreme guardian of phosphatidylethanolamine (PE). The GPX4 gene produces an enzyme that neutralizes free radicals right inside lipid membranes.
- In healthy cells: it functions normally, preventing PE peroxidation and death by ferroptosis.
- In drugs: many new compounds target GPX4 only in tumors, leading to oxidative collapse.
2. NRF2 (The master regulator)
This gene is like a stress thermostat. When it detects free radicals, it activates a cascade of over 200 antioxidant genes.
- Protection: NRF2 stimulates the production of glutathione, which GPX4 uses to repair membranes.
- Selectivity: Healthy cells use NRF2 in a balanced way. Many tumors, however, "hijack" NRF2 to become immortal; researchers are studying how to target this specific addiction.
3. FSP1 (Ferroptosis Suppressor Protein 1)
Recently discovered, this gene acts as a backup system. Even if GPX4 is blocked, FSP1 can protect cell membranes by reducing Coenzyme Q10, which acts as a powerful “radical-scavenging” antioxidant.
Why is this important for therapy?
The modern strategy is no longer “kill everything that grows” (like the old chemotherapy), but:
- Analyze the patient's genetic profile.
- Identify which shield (GPX4 or FSP1) the tumor is using to protect its phosphatidylethanolamine.
- Strike that specific shield, trusting that healthy cells have alternative, more robust metabolic pathways to remain intact.
4. ETNK1 (Ethanolamine Kinase 1)
ETNK1 gene is the link between two fundamental metabolic processes on either side of the energy balance that regulates cellular homeostasis: it is the enzyme responsible for the transformation of ethanolamine into phosphoethanolamine, the first step in the metabolic pathway that leads to the creation of phosphatidylethanolamine (PE), which is regulated by the function of the GPX4 gene responsible for the formation of reduced glutathione, which, in turn, derives from folate metabolism. This means that people with a homozygous MTHFR gene mutation in the C677T conformation are more susceptible than average to tumor formation if exposed to environmental factors that stimulate hyperhomocysteinemia.
Here’s how this gene malfunction opens the door to carcinogenesis:
1st. The “assembly line” block
When the ETNK1 gene mutates (often “gain-of-function” or “loss-of-function” mutations, depending on the tumor, but frequently studied in myeloid diseases), the enzyme malfunctions, leading to:
- Substrate accumulation: untransformed ethanolamine accumulates.
- Product deficiency: phosphoethanolamine levels decrease, and thus, PE levels in membranes.
2nd. Fragile membranes and free radicals
As we have seen, PE is structural. If ETNK1 is mutated, the cell produces “defective” membranes. These membranes are much more vulnerable to free radical attack because they lack the correct lipid balance between phosphatidyl ethanolamine and the other two structural molecules, phosphatidylserine and phosphatidylcholine. An unstable membrane facilitates the reception of protumor signals.
3rd. Connection with Mitochondria
PE produced via the ETNK1 pathway is essential for mitochondrial health. Mutations in this gene lead to:
- Mitochondrial dysfunction: mitochondria "leak" electrons, dramatically increasing internal free radical production.
- Metabolic stress: the cell enters a state of perpetual alert that favors further genetic mutations that increase susceptibility to carcinogenesis
A person with germline (hereditary) or somatic (acquired) mutations in ETNK1 exhibits “basic metabolic fragility.” This means:
- Reduced repair capacity: if free radicals (from pollution, diet, or smoking) damage membranes, the cell lacks enough phosphoethanolamine to rapidly rebuild them.
- Susceptibility to ferroptosis or incorrect apoptosis: blood cells (where these mutations are typical, as in Chronic Myelomonocytic Leukemia) are unable to properly regulate their own death, leading to abnormal proliferation.
ETNK1 mutations should therefore be considered “drivers,” or motors that push a normal cell to become cancerous by radically altering the way it processes fats and energy.
Ivermectin
Ivermectin is not currently an approved drug for the treatment of cancer in humans, but there are excellent evidences, especially in the case of glioma or neuroblastoma, in the observational (Phase 1) experimental phase.
The main mechanisms through which ivermectin appears to act on human cancer cells are:
1. Inhibition of signaling pathways (WNT and YAP)
Cancer cells often “hijack” certain cellular communication pathways to grow uncontrollably. Ivermectin appears to intervene by blocking:
- The WNT/beta-catenin pathway: this pathway is often overactive in colon cancers. Ivermectin inhibits the beta-catenin protein, slowing cell replication.
- The YAP/TAZ pathway: crucial for the growth of many solid tumors; the drug prevents its activation.
2. Induction of apoptosis and autophagy
Ivermectin can push cancer cells into “cell suicide” through:
- Oxidative stress: increases the production of reactive oxygen species (ROS), which damage the tumor cell internally.
- Apoptosis: activates caspases, enzymes that dismantle the cell from within.
3. Effect on the tumor microenvironment and immunogenicity
One of the most interesting discoveries concerns so-called immunogenic cell death ( ICD ). Ivermectin not only kills the cell, but also releases chemokines that help T lymphocytes recognize and attack the tumor.
4. Inhibition of cancer stem cells
Cancer stem cells are often resistant to traditional chemotherapy and cause tumor recurrence. Some studies indicate that ivermectin may selectively target these cells, making the tumor less likely to recur.
Main challenges
Despite these promising mechanisms, there are significant obstacles:
Dosage: Many of the antitumor effects observed in the laboratory require drug concentrations much higher than those safe for humans as an antiparasitic. This may depend on the conditions of the cellular microenvironment, and therefore, the presence of substances capable of rebalancing it may be necessary, in vivo and not just in vitro or in silico. We use ivermectine 0,3 mg/ml for 8 inhalations a day.
Bioavailability: It’s difficult to deliver the drug exactly where it’s needed (inside the tumor mass) without causing toxicity in other organs. For this reason, we’re testing a liposomal spray composition that can be inhaled directly through the nostrils and cross the blood-brain barrier. In summary: Ivermectin acts as a “multi-target” drug that interferes with the metabolism and communication of cancer cells, but large clinical trials are needed to confirm whether these actions are effective and safe in human patients. We are promoting the use of ivernectin because we had seen a good tumor response.
Project 1
Chronic sub-lethal oxidative stress as a mutagenic cofactor in ETNK1 and consequent gain of function of phosphoethanolamine metabolism in tumor cells: use of antioxidant compounds to restore redox and cellular metabolic balance.
Notice
The following is a parallel project, born following evidence of renal failure during an infusion in a patient who, only later, was discovered to carry a mutated and therefore nonfunctional SUOX gene. The situation was resolved by withdrawing NAC for a couple of weeks and subsequently readjusting it to a minimum dose of 150 mg.
The project is summarized in the parts that are specific to the original work and have been added to this document only for completion and further study. Biochemical pathways
1. ROS and binding to phosphatidylethanolamine
ROS (•OH, O2•–, H2O2 Fenton pathway) potentially target the double bonds of the polyunsaturated acyl chains (PUFA) of phosphatidylethanolamine → formation of lipid radicals → LOO• → LOOH. • Secondarily: formation of reactive aldehydes (4-HNE, MDA).
The ethanolamine group of PE can form adducts with secondary aldehydes (e.g., 4-HNE), but this is a “downstream” effect of peroxidation, not the starting point.
2. ROS and membrane synthesis
There is no evidence that ROS directly regulate PE synthesis via structural interaction with the molecule, but they can interfere with PE synthesis. It is regulated by:
- CDP-ethanolamine pathway (Kennedy pathway)
- Decarboxylation of phosphatidylserine in Mitochondria (PSD)
- Regulation by mTOR, AMPK, energy status
ROS can indirectly modulate these pathways through:
- Activation of NRF 2
- Inhibition of redox-sensitive cysteine-containing enzymes
- Alteration of mitochondrial metabolism
3. ROS, PE, and oncogenesis: the critical point is elsewhere
The strongest documented link (Q1 journals, Nature, Cell, Cancer Cell, last 10–15 years, highly cited) concerns ferroptosis.
PE containing PUFAs (e.g., AA-PE, AdA-PE) is oxidized by:
Lipoxygenase (ALOX)
Free iron (Fenton reaction) → formation of specific Pe-OOH → trigger of ferroptosis.
Here, PE is central, but as an oxidizable lipid substrate, not as a direct regulator of mitosis.
4. ROS and cancer mitosis: real mechanism
ROS-induced tumor proliferation occurs through the activation of redox-sensitive and proliferative pathways:
- MAPK
- PI3K/AKT
- NF-κB
- HIF-1α
RATIONALE: Phosphatidylethanolamine (PE) is a key phospholipid in biological membranes (especially mitochondrial membranes), and its synthesis is finely regulated by energy and cellular stress conditions, involving the mTOR, AMPK, and NRF2 signaling pathways.
Synthesis of Phosphatidylethanolamine (PE)
PE constitutes 20-30% of membrane lipids in mammalian cells. Its synthesis occurs primarily in the endoplasmic reticulum (ER) and mitochondria through three main pathways:
- CDP-ethanolamine pathway (Kennedy Pathway): This is the primary pathway in the endoplasmic reticulum, where ethanolamine is converted to CDP-ethanolamine and then combined with diacylglycerol (DAG).
- Phosphatidylserine (PS) decarboxylation: This occurs in the mitochondria, where the enzyme PS decarboxylase (PSD) converts phosphatidylserine to PE. This pathway is crucial for mitochondrial biogenesis.
- Ca2+-dependent headgroup exchange: Free ethanolamine replaces the headgroup of other phospholipids (PS or PC).
Role of mTOR, AMPK, and NRF2 in regulating β-sphatidylethanolamine synthesis
These players regulate PE synthesis primarily by responding to nutritional status and oxidative stress (related to mitochondrial function).
mTOR (Mammalian Target of Rapamycin) - Anabolism and Growth:
- Role:When active (in response to high nutrients), mTOR stimulates cell growth and anabolism, including lipid synthesis.
- Interaction with PE: mTORC1 can promote the synthesis of membrane lipids, including PE, to support cell expansion. However, excessive mTOR is associated with insulin resistance.
AMPK (AMP-activated Protein Kinase) - Energy Sensor:
- Role: Activated under low-energy conditions (low ATP, high AMP) to inhibit anabolism and promote catabolism, often by inhibiting mTOR.
- Interaction with PE: AMPK inhibits fatty acid synthesis but can activate phospholipid remodeling pathways to maintain cellular homeostasis in stressful situations. It also promotes mitophagy (removal of damaged mitochondria), a process that requires fine-tuning of mitochondrial PE.
NRF2 (Nuclear Factor Erythroid 2-Related Factor 2) - Oxidative Stress:
- Role: It is the main regulator of the antioxidant response, activated by oxidative stress.
- Interaction with PE: PE is abundant in mitochondria, where ROS production is high. Adequate PE synthesis (via PSD) is necessary for mitochondrial health. NRF2, activated by AMPK under stressful conditions, can promote the expression of antioxidant genes that protect the integrity of PE-rich membranes.
Summary of Biochemical Relationships
- AMPK acts as a master switch: when energy is low, it inhibits mTOR (reducing general protein/lipid synthesis) and activates NRF2 to protect membranes.
- PE synthesis in the mitochondrion (PSD pathway) is essential during mitochondrial biogenesis (promoted by growth conditions/mTOR or energy restoration), while its synthesis in the ER (Kennedy pathway) is more dependent on the availability of precursors.
- PE is essential for autophagy (autophagosome formation), a process regulated by the mTOR/AMPK complex.
- Inactivation of redox-sensitive phosphatases (PTEN) → Increased proliferative signaling
- Oxidative DNA damage → Driver mutations
- Alteration of cell membrane fluidity → clustering of tyrosine kinase receptors (EGFR). PE plays an indirect role when, due to permanent oxidative stress, peroxidation alters membrane curvature, thus altering lipid raft domains and vesicular trafficking.
- Critical conceptual point: membrane synthesis is regulated primarily by mTOR and lipid metabolism, not by local PE modifications.
Phospholipid oxidation is most often:
- cytotoxic
- pro-ferroptotic
- pro-apoptotic
To become pro-oncogenic, a moderate but chronic level of ROS is required, and this occurs during permanent oxidative stress.
6. Most plausible hypothesis
Excess ROS → Increased PE-PUFA peroxidation to a pathological level; this causes alterations in membrane microdomains, resulting in changes in RTK clustering, leading to chronic PI3K/AKT activation with increased lipogenesis (SREBP1). Increased lipid synergy stimulates the ETNK1 enzyme to produce phosphoethanolamine, inducing tumor-promoting metabolic reprogramming with lipid remodeling.
Clear conclusion: the oxidation of PE PUFA chains by excess ROS, following persistent oxidative stress, may indirectly contribute to proliferative signaling through alterations in membrane phospholipid folding and redox-dependent pathways.
Increased ROS lead to a metabolic link with parallel activation of the CDP-ethanolamine or CDPcholine pathway, for example, through the following series of reactions:
- Ethanolamine kinase (ETNK1/ 2) Ethanolamine + ATP → Phosphoethanolamine + ADP
- CTP: phosphoethanolamine cutidyl transferase (PCYT 2) Phosphoethanolamine + CTP → CDP-ethanolamine + Ppi
- Ethanolamine phosphotransferase (EPTI/SELENOI) CDP-ethanolamine + DAG → Phosphatidylethanolamine (PE) + CMP
None of these reactions transfer electrons, so we can’t imagine a redox mechanism, yet phospholipid activation, in turn, leads to ROS accumulation, a self-perpetuating mechanism in tumors. There are four plausible mechanisms, all of which could be effective, as they are energetically synergistic:
Mechanism 1 — Increased mitochondrial blood flow Activation of phospholipid synthesis causes:
↑ ATP consumption
↑ NADPH requirement (for related lipogenesis)
↑ β-oxidation or compensatory glycolysis
This increases the flux in the electron transport chain (ETC).
Increased flux = greater probability of electron leakage from complexes I and III and formation of superoxide (O2•–) Therefore:
Lipogenic activation → increased respiration → increased mitochondrial ROS.
It is not the Kennedy pathway that produces ROS, but the energy demand that increases electron leakage.
Mechanism 2 — Alteration of mitochondrial membrane composition
PE is essential in the mitochondrial inner membrane. Quantitative or qualitative alterations in PE cause modification of the curvature of the mitochondrial outer membrane, altering the organization of the respiratory complexes by deviating the orientation of the magnetic omenta, hindering electron tunneling between the outermost bond levels, destabilizing the redox supercomplexes necessary to ensure the maintenance of the electrical potential of the respiratory chain, which acts as the motor for transporting electrons from pyruvate to oxygen. This can increase the inefficiency of the ETC → ↑ ROS production. This phenomenon has been documented in models of mitochondrial PE depletion, so it is plausible.
Mechanism 3 — Interaction with ferroptosis
PE containing PUFAs are substrates for peroxidation:
PE-PUFA + Fe²⁺ → PE-OOH
Here, ROS are not generated by the Kennedy pathway, but the increase in PE-PUFA provides an oxidizable substrate and, in the presence of free iron, increases radical propagation, increasing the vulnerability of the mitochondrial membrane.
Mechanism 4 — NADPH and redox balance
Global lipogenesis is linked to the pentose phosphate pathway as well as two other phenomena:
- malic enzyme
- cytosolic IDH
If NADPH is drained by lipid biosynthesis, the result is ↓ antioxidant capacity (GSH/thioredoxin) → secondary ROS increase.
4. When the pentose phosphate pathway can reduce ROS A correct level of mitochondrial PE:
- stabilizes complex IV
- improves respiratory efficiency
- reduces electron leakage
Therefore, the pathway can also reduce ROS if it corrects a PE deficiency.
Logical summary
The Kennedy pathway does not generate ROS by direct redox mechanism but can modulate ROS indirectly via:
- energy metabolism
- mitochondrial organization
- vulnerability to peroxidation
- NADPH balance
Operational recommendation during experimentation
1. Quantify specific PE-OOH (targeted lipidomics LC-MS).
2. Evaluate the correlation with:
- AKT phosphorylation
- ERK phosphorylation
3. To do this, we can use lipoxygenase inhibitors or iron chelators and evaluate whether blocking PE peroxidation reduces proliferation.
If proliferation decreases independently of DNA damage, then we have a functional lipid signal.
To clarify the mechanism, we need to:
- Measure oxygen consumption (Seahorse).
- Measure NADPH/NADP⁺ concentration.
- Evaluate the presence of mitochondrial vs. cytosolic ROS.
- Analyze PE-PUFA lipidomics.
- Use iron chelators to distinguish the ferroptotic component.
Only in this way can a clear causal link be established.
I’m developing a rigorous experimental design to test whether activation of the Kennedy pathway, in its phosphoethanolamine-producing branch, leads to an increase in ROS, and through what mechanism.
1. Hypotheses to be discriminated we must separate four possibilities:
H1: energetic effect ↑ Kennedy flux → ↑ ATP demand → ↑ ETC flux → ↑ mitochondrial ROS
H2: Effect on mitochondrial composition Alteration of mitochondrial PE → destabilization of supercomplexes → ↑ electron leak
H3: PE-PUFA peroxidation effect↑ PE-PUFA → ↑ peroxidizable substrate → ↑ radical propagation
H4: redox effect with NADPH depletion ↑ lipid biosynthesis → ↓ available NADPH → ↓ antioxidant capacity → ↑ ROS
My goal is to distinguish which of these is dominant.
Experimental system
Cell line
- A proliferative tumor cell line (e.g., HCT116 or A549)
- A non-tumor cell line (physiological control)
Genetic manipulations
- ETNK1 Overexpression
- ETNK1 Knockdown (shRNA/CRISPRi )
- PCYT2 Overexpression
- Empty Vector Control
A rescue experiment is also needed.
Primary endpoints
A. Total and compartmental ROS
- DCFDA (cytosolic ROS)
- MitoSOX (mitochondrial ROS)
- HyPer redox sensor (better, compartmentalized) Fundamental distinction:
If ROS ↑ mitochondrial only → supports H1 or H2.
B. Bioenergetics (Seahorse) Measure:
- Basal OCR
- Maximum OCR
- Proton leak
- Spare respiratory capacity
If proton leak ↑ → supports H2.
If global OCR ↑ without increased leakage → supports H1.
C. NADPH/NADP⁺ ratio
Enzymatic cycling assay.
If ↓ NADPH with ↑ Kennedy → supports H4.
D. Lipidomics LC-MS
Quantify:
- Total PE
- PE-PUFA (AA-PE, AdA-PE)
- PE-OOH
If ↑ PE-OOH → supports H3.
Discriminating pharmacological interventions
To separate the mechanisms:
To verify H1 (respiratory flow):
- Oligomycin
- Rotenone
- Antimycin A
If ROS normalize → ETC dependence. To verify H3 (ferroptosis-like):
- Deferoxamine (iron chelator)
- Liproxstatin-1
If ROS significantly ↓ → iron-dependent component.
To verify H4:
- Supplementation with NADPH precursor (e.g., ribose-5-phosphate via PPP stimulation)
- FASN inhibition
Mitochondrial structural analysis
Electron microscopy:
- Cristae density
- Disorganization of internal membranes Structural alterations → H2 support.
Timeline
Fundamental:
- 6 hours
- 24 hours
- 48 hours
If ROS increase before lipid alterations → metabolic cause.
If ROS increase after PE-PUFA accumulation → peroxidative cause (most accepted hypothesis)
Functional output
- Proliferation (BrdU)
- Cell cycle analysis
- Ferroptosis markers (GPX4, lipid ROS)
Logical interpretation
Osservation main mechanism
| ↑ ROS mito + ↑ proton leak | H2 |
| ↑ ROS mito + ↑ OCR | H1 |
| ↑ PE-OOH + iron chelant | H3 |
| ↓ NADPH without ↑ OCR | H4 |
Methodological critical issues
- DCFDA is nonspecific.
- MitoSOX can be artifactual.
- Artificial overexpression can create nonphysiological stress.
- Tumor cells already have altered redox response. Replication in at least two lineages is required.
Strategic conclusion
Do not assume that the Kennedy pathway generates ROS.
Test first:
- ROS compartmentalization.
- Respiratory flow.
- NADPH status.
- PE peroxidation.
Only then can we infer causality.
Operational recommendation
Start with:
- ETNK1 overexpression
- Seahorse
- MitoSOX
- PE-PUFA targeted lipidomics
This is the minimum package to understand whether we are observing a bioenergetic or lipid phenomenon.
At this point, in the case of tumors of the proliferative lineage of the bone marrow (leukemia, lymphoma, myeloma, etc.), the use of a substance that inhibits the uncontrolled proliferation of phosphoethanolamine by inhibiting the ETK1 enzyme when it is in its mutated and hyperactive form can be studied. This alone is certainly not sufficient, but if combined with regulation of the ionic balance of the water clusters that form the substrate for electron transport in the cytoplasm (state 4 water), thanks to the action of an antioxidant solution, this can act, as it normally does, by recalibrating the direction of the electromagnetic moment of the cytoplasmic magnetic field, “forcefully” restoring the mtochondrial membrane to its energetically favorable conformation, allowing the correct potential differential between the two ends of the oxidative phosphorylation chain to be maintained and, therefore, electron tunneling with the formation of physiological ATP. The cell does not react to pseudohypoxia and returns to its most energetically favorable state, i.e., the basal state, either by “reversing” the tumor or by promoting the translocation of phosphoethanolamine to the outer membrane, triggering a cytokine reaction that attracts CD8+ T lymphocytes and, therefore, the apoptosis of tumor cells alone. This does not conflict with chemotherapy, which essentially do the same thing; rather, it has a synergistic effect, increasing their efficiency, reducing adverse effects, and promoting faster recovery and a significant reduction in mortality.
The proposed research goes in both directions: calibrating antioxidant levels through intravenous drips containing glutathione, N-acetyl cysteine, and vitamin C (CRRT (Cellular Redox Rebalance Therapy) method) and studying molecules that inhibit the mutated ETNK1 enzyme.
Research project proposal: Randomized, controlled, adaptive study on the timing and personalized dosing of antioxidants as adjuvants to cytotoxic chemotherapy: impact on toxicity, antitumor efficacy, and clinical outcomes.
Biochemical Rationale
Oxidative stress and degenerative diseases: Persistent oxidative stress is the basis of all degenerative diseases. Our body, which is attacked daily by oxidants, is able to restore and balance the Ox/Red ratio to 1 thanks to the action of specific molecules capable of reducing free radicals, which are normally formed as a product of cellular metabolism and play important regulatory roles in cell function. However, when the stressful stimulus lasts beyond the cell’s recovery time, primarily through the action of glutathione, this ratio cannot be restored, and the cell experiences persistent oxidative stress. By experiencing persistent oxidative stress, the methylation process and the glutathione cycle are disrupted, and by disrupting the glutathione cycle, the molecular shield that protects us from environmental carcinogens— approximately 10,000, but 400 transforming ones for humans (according to Professor Henry Busch) —is disrupted.
I start from some well-known assumptions, specifically:
- In all patients with degenerative diseases and many other pathologies, the relationship between the reducing and oxidizing systems is imbalanced due to a deficiency in a molecule, GSH, which keeps the entire reducing system in a reduced state.
- The reducing system derives from the cooperation of GSH with the oxidoreductive activity of ascorbate.
- Under normal conditions, GSH is continuously regenerated through a sequence of reactions involving two enzymes: glutathione peroxidase and glutathione reductase.
Some chemical compounds, many of which are structurally similar to the substrates, have the property of decreasing enzymatic activity to the point of blocking it completely (competitive inhibition) or of binding chemically to the enzyme or enzyme-substrate complex, preventing its cleavage (noncompetitive inhibition). Inhibition is generally reversible: by increasing the concentration of the substrate (competitive inhibition) or by removing the inhibitor (noncompetitive inhibition). It is known that various types of metabolites, generally chemically different from the substrates, are capable of increasing or decreasing the activity of certain ions called allosteric ions (in biochemistry, an enzyme whose activity is regulated by a particular effector molecule, generally of small molecular weight, which can bind to a site other than the active site, modifying the behavior of the enzyme itself), usually trace elements such as manganese, calcium, iron, and zinc.
In non-competitive inhibition, the inhibitor alters the enzyme. This explains the mechanism of many toxic substances (e.g., heavy metals such as Hg, Pb, etc.) which inhibit many enzymes. Some of these substances, given the chemical groups with which they react, are used in vitro to reveal the enzyme’s concentration. Thus, for example, if the enzyme is inhibited by substances that react with –SH groups, it means that these are necessary for its action. It reacts, for example, with: with the – SH groups iodoacetamide: RSH + ICH2CONH2 = RS CH2CONH2 + HI, but other xenobiotic elements, exogenous and/or endogenous, react with the thiol groups to be neutralized. The efficiency of the lymphocyte immune system is also linked to the cellular redox system, so if one becomes unbalanced, the other becomes unbalanced.
Summary of vital cycles involved in oncogenesis
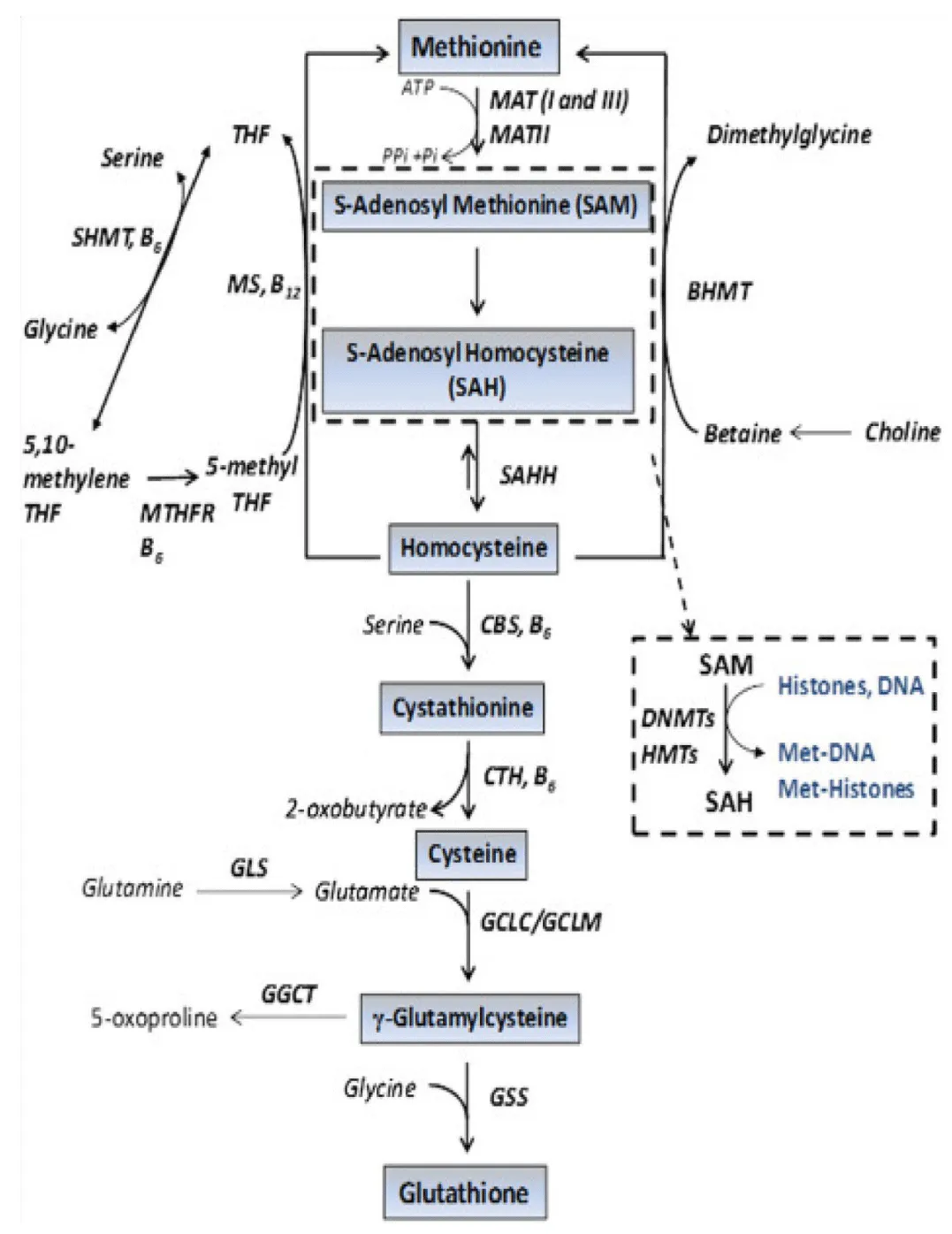
Therefore, the cellular defense mechanism against oxidants can generally be summarized as follows:
Ascorbate + ag, oxidant ◊ dehydroascorbate + H2O2
Dehydroascorbate + 2 GSH ◊ Ascorbate + GSSG
H2O2 + 2GSH ◊ 2 H2O + GSSG
GSSG + NADPH + H+ ◊ 2GSH + NADP+
The lack of GSH availability, due to mutations in the SOD or catalase genes or to GPX4 malfunction, is also directly responsible for significant alterations in red blood cells, especially affecting the integrity of the membrane and hemoglobin, but, above all, causes severe systemic oxidative stress. In this situation, if the cellular homeostatic redox balance is not reestablished, no type of targeted therapy will ever be effective definitively against degenerative diseases, where relapses are often common.
It is also counterproductive, and sometimes harmful, to administer supplements without first assessing the biochemical system through blood tests. Therefore, it is always best to check blood levels before taking supplements that can also act as pro-oxidants. In cases of drug resistance, which can be determined by verifying the function of cytochrome P450 enzymes or by the presence of hypofunctional polymorphisms in the genes that produce them, it is essential to assess the redox status with appropriate biochemical tests and administer antioxidant therapy via IV drip to reactivate the cellular homeostatic redox system before chemotherapy (typically, there are reports of protocols applying it 48-72 hours before and after the infusion). Studies on the importance of pharmacogenomics are well known today, but the most important in Italy is the Rome Trial: from blood sampling to molecular targeting, promoted by La Sapienza University of Rome, the ISS, and the Institute for Personalized Medicine. It states, “Subsequently, through a specific genomic evaluation, it will be possible to identify patients who have a greater chance of responding to immunotherapy, laying the foundation not only for molecularly targeted drugs, but also for personalized care, developed on the basis of a genomic profile.” The author shares this line of thought and therefore suggests testing cancer patients’ ability to metabolize drugs. This can be done with simple steps that should be part of routine pre-chemotherapy practice, also because they appear to allow for the personalization not only of the dosage, but also of the type of chemotherapy that offers the greatest therapeutic benefit. This can be done in two main ways:
1. Therapeutic Drug Monitoring (TDM)
The most direct method involves measuring the plasma concentration of a drug the patient is already taking.
- What is measured: The ratio between the administered dose and the concentration of the drug (and its metabolites) in the blood.
- How to interpret it:
- Levels that are too high suggest a slow metabolism (Poor Metabolism). The risk is toxicity.
- Levels that are too low suggest an ultra-rapid metabolism (Ultrarapid Metabolism). The risk is therapeutic ineffectiveness.
- Limitation: This test is specific to the drug being used and does not necessarily indicate "why" the metabolism is altered (it could be due to interference with other drugs, diet, or liver disease).
2. Testing with Probe Substrates (Phenotyping)
To test the activity of a specific enzyme (e.g., CYP2D6 or CYP3A4) before initiating major therapy, so-called “probe drugs” are used.
- Procedure: A standard dose of a harmless substance that is metabolized exclusively by a specific cytochrome C190 enzyme is administered.
- Analysis: After a specified time, a blood sample is drawn to calculate the Metabolic Ratio (MR) as the ratio of the initial drug concentration to that at a specified time.
- Common examples:
- Dextromethorphan: to test for CYP2D6.
- Caffeine: often used for CYP1A2.
- Midazolam: for CYP3A4.
If the above tests indicate an abnormality (e.g., an unbalanced metabolic ratio), pharmacogenetics is performed. Through a simple blood sample (from which leukocyte DNA is extracted), single nucleotide polymorphisms (SNPs) are searched for. Those of particular interest are the genes responsible for the metabolism of the most common chemotherapy drugs.
Chemotherapy metabolism is an extremely complex process involving several enzymes of the Cytochrome P450 (CYP) system. Identifying which enzyme is predominant is crucial, as genetic variations (polymorphisms) in these genes can lead to excessive toxicity or poor treatment efficacy.
Main cytochromes involved in the metabolism of oncology drugs:
1. CYP3A4 and CYP3A5 (the most important)
They are responsible for the metabolism of over 50% of currently used drugs, including many chemotherapy drugs.
Drugs involved: Taxanes (Docetaxel, Paclitaxel), Vinca alkaloids (Vincristine, Vinblastine), Tyrosine kinase inhibitors (Imatinib, Gefitinib), Irinotecan, and Etoposide.
Note: These enzymes are highly susceptible to drug interactions (many drugs can “block” or “accelerate” them).
2. CYP2D 6
It is a highly polymorphic enzyme (many genetic variants exist in the population).
Key drug: Tamoxifen (hormone therapy for breast cancer). CYP2D6 converts tamoxifen into its active metabolite, endoxifen. If a patient is a “poor metabolizer” for CYP2D6, therapy may be ineffective.
Other drugs: Some antiemetics used to counter chemotherapy nausea (such as ondansetron).
3. CYP2C 9
Drugs involved: Cyclophosphamide and ifosfamide (partially), but it is also essential for supportive drugs such as warfarin or some NSAIDs.
4. CYP2C 19
Drugs involved: Cyclophosphamide (activation), thalidomide, and some proton pump inhibitors often administered concurrently with chemotherapy.
5. CYP1A 2
Drugs involved: Flutamide and some drugs activated or inactivated by cigarette smoking (which induces this enzyme).
Other useful polymorphisms, when combating side effects or for the use of first-line drugs following the adverse effects of chemotherapy, are:
- CYP2D6: involved in the metabolism of antidepressants, antipsychotics, and opioids.
- CYP2C19: crucial for clopidogrel (antiplatelet) and proton pump inhibitors.
- CYP2C9: crucial for warfarin (anticoagulant) and NSAIDs.
Summary of the approach
| Fase | Tipo di Analisi | Obiettivo |
| 1. Screening | TDM | Check if the metabolism is actually altered (Phenotype). |
| 2. In-depth analysis | Pharmacogenetic PANEL |
Identify allelic variants that explain the defect (Genotype). |
| Note: The phenotype may change over time due to the action of some chemotherapeutic agents that “induce” or “inhibit” enzymes. Furthermore, the elevated oxidative stress induced by chemotherapy produces a large amount of ROS, which, among other effects, has epigenetic properties and, therefore, causes enzyme dysfunction by inhibiting DNA methylation. Always keep this risk in mind. |
||
Basic CRTT therapy scheme for the treatment of degenerative disease
Fluor drug inection (prodigy) one every 24 hours composition of the inectable solution:
- 500ml 5% glucose solution (in case of hyperglycemia, use saline. Once blood levels have returned, return to glucose) only in the presence of cachexia, otherwise use saline.
- 3 vials of 600 mg of GSH
- 2 vials of 300 mg of N-acetylcysteine.
- 1 vial of Ranidil, 50 mg (5 ml).
- 1 vial of 1 gram of ascorbic acid.
Notice:
- In cases of neutrophilia, vitamin C should be increased to 3 grams with a slow infusion.
- In cases of high COVID IGG levels above 10,000 or in the presence of adverse events with neurological damage.
- In cases of high COVID IGG levels above 10,000 or in the presence of adverse events with neurological damage, a "strengthened" version of the therapy is used: 500cc + 3 x 600 mg GSH + 3 x 300 mg NAC + 2 grams of vitamin C slow infusion. One every 24 hours.
Important: Ascorbates bind the oxidizing agent, and the thiol group of GSH expels it, while glutathione reabsorbs it via γ-glutamyltransferase, located in the brush border.
Based on case reports, this therapy may allow for a reduction in chemotherapy adverse events and facilitate recovery.
Evidence from the literature
I’d conducted extensive literature searches for comparisons with similar therapies used as adjuvants in cancer treatment and highlighted several critical issues:
- Timing and sequence have not been systematically standardized in current trials.
- Robust oncology endpoints such as OS/PFS are rarely primary outcomes in antioxidant trials.
Key points from peer-reviewed evidence
- There is no robust clinical evidence that the use of antioxidants is detrimental or reduces the efficacy of chemotherapy/radiotherapy in available randomized trials.
- There are signs of reduced toxicity, especially for glutathione and NAC in some settings, but further research is needed, especially methodological ones.
- There are no well-defined clinical studies testing the timing/strategic dosing of antioxidants as timed adjuvants to improve clinical outcomes in oncology.
Therefore, a protocol is proposed to address this gap
- The reviews conclude that the use of these antioxidants in oncology cannot be definitively recommended or ruled out without further high-quality studies. Without any particular pretensions, the proposed study aims to explore the intracellular biochemical mechanisms that influence carcinogenesis.
- The use of individual components of CRRT (Cellular Redox Rebalance Therapy) is widely used in pharmacology (e.g., TAD 600, Fluimucil, vitamin complexes) to reduce the toxicity of chemotherapy.
Based on these results and the above, the following project is proposed. Initially, it will be an observational study, and, once any adverse effects have been verified and the relative and absolute efficacy have been calculated, it will progress to a Phase 2 study.
Project 2
Randomized, controlled, adaptive study on the timing and personalized dosing of antioxidants as adjuvants to cytotoxic chemotherapy: impact on toxicity, antitumor efficacy, and clinical outcomes.
Scientific rationale
Cytotoxic chemotherapy exerts part of its efficacy through oxidative stress and DNA damage, but also induces dose-limiting toxicities in healthy tissues. The existing clinical literature on antioxidants during chemotherapy is characterized by:
- heterogeneity of molecules, doses, and timing;
- lack of pharmacokinetic/pharmacodynamic (PK/PD) stratification;
- often surrogate or secondary clinical endpoints;
- insufficient statistical power;
- lack of temporal separation between the cytotoxic phase and the tissue recovery phase.
Central hypothesis: Antioxidant administration temporally decoupled from the cytotoxic peak and personalized based on PK/PD and oxidative stress biomarkers can reduce toxicity without reducing antitumor efficacy.
Testable Hypotheses (end points)
- H1 (safety): Post-cytotoxicity peak administration does not reduce ORR, PFS, or OS compared to control.
- H2 (clinical efficacy): The antioxidant arm significantly reduces grade ≥2 toxicity (CTCAE v5.0).
- H3 (mechanistic): The reduction in toxicity correlates with biomarkers of systemic and tissue oxidative stress, without reducing tumor oxidative damage.
Most clinical trials do not incorporate standardized timing/dosing designs to maximize the desired protection vs. cytotoxic effect profile.
Systematic reviews find mixed results: some studies show a reduction in toxicity, others no effect, and some report potential interference with tumor efficacy.
Common methodological limitations found in previous studies:
- Small sample sizes, different chemotherapy regimens, inconsistent antioxidant formulations, and variable primary clinical outcomes.
- Administration timing rarely accurately stratified with respect to peak pharmacokinetics of chemotherapy agents in available trials.
Overall assessment
There is no strong clinical consensus based on high-quality RCTs defining the safety, efficacy, and optimal timing/dosing parameters for antioxidants during chemotherapy.
Many highly cited reviews remain cautious or negative regarding the routine use of supplemental antioxidants during cytotoxic treatments, precisely because of the potential risk of interfering with ROS-mediated tumor killing.
There are no robust data from high-quality RCTs defining an antioxidant + chemotherapy protocol with a clear reduction in toxicity without compromising antitumor efficacy. Despite all this, the data I have come across and other literature suggest that in some selected settings, antioxidants can alleviate the toxicity of chemotherapy; in others, they can reduce efficacy, increase recurrence, or worsen survival. The optimal timing (e.g., far from the pharmacokinetic peak) is necessary by highquality RCTs.
Any use of antioxidants during chemotherapy must be evaluated in a strictly controlled clinical setting by oncologists, biologists, biochemists, and chemists, with access to specific therapeutic protocols, and ideally, be included in well-designed clinical trials that fall within the scope of this trial (e.g., with rigorous endpoints and stratification of dosage and timing).
Indeed, most of the clinical trials conducted to date and available in the literature reveal serious methodological flaws and, in some cases, scientific inconsistencies. For example, most studies do not incorporate standardized timing/dosage designs to maximize the desired protection vs. cytotoxic effect profile. Systematic reviews, as mentioned, find mixed results: some studies show reduced toxicity, others no effect, and some report potential interference with tumor efficacy.
It is the author’s opinion that the main methodological flaws can be summarized in five main areas:
- small sample sizes;
- different chemotherapy regimens;
- non-homogeneous antioxidant formulations;
- variable primary clinical outcomes.
- administration timing rarely accurately stratified with respect to the pharmacokinetic peaks of the chemotherapy drugs in available trials.
Conclusion derived from a review of the literature in indexed journals (Q1, Q2)
Despite methodological differences and necessarily divergent conclusions given that the studies are not guided by a similar protocol, which is why, although the controlled clinical literature indicates that the use of antioxidants as adjuvants in chemotherapy/radiotherapy is not intrinsically harmful, while other literature claims that it may reduce some adverse effects, there is no robust evidence that they improve survival outcomes or tumor response. This is not because this is not the case, since biochemistry teaches and demonstrates precisely the opposite (as explained at the beginning), but because methodological inconsistencies make the current evidence fragile, heterogeneous, and incapable of establishing timing, dosage, and mechanisms (such as epigenetic or redox) with sufficient rigor.
Well-designed prospective clinical trials, with rigorous definitions of antioxidant, dose, timing, and clear oncological endpoints, are needed to establish a reliable and safe clinical role for these agents as adjuvants. This is the main purpose of the project.
The role of oxidative stress in cancer development and progression has attracted increasing attention in recent years. Persistent oxidative imbalance may contribute to DNA damage, mitochondrial dysfunction, chronic inflammation, and abnormal activation of signaling pathways involved in carcinogenesis.
The present proposal integrates current knowledge regarding phospholipid metabolism, ETNK1 dysfunction, ferroptosis, and antioxidant regulation into a broader biochemical framework. The proposed CRRT approach aims to support restoration of cellular redox balance while potentially reducing chemotherapy-associated toxicity.
Although several studies have investigated the use of antioxidants during chemotherapy, available evidence remains inconsistent due to methodological variability among published trials. Factors such as antioxidant dosage, timing of administration, patient selection, and pharmacogenomic differences may significantly influence clinical outcomes.
The proposed study may contribute to a better understanding of personalized redox-based therapeutic strategies and help clarify the relationship between oxidative stress, phospholipid metabolism, and tumor biology.
Operational guidelines during the observational study
- Do not use high-dose antioxidants empirically outside of controlled clinical trials, especially when combined with anticancer therapy. Studies with controlled dose escalations and under close medical observation and monitoring of the patient's vital signs are preferred.
- When considering antioxidants as adjuvants, there must be specific medical supervision by a specialist (e.g., oncologist) within a precise planning that defines dose, timing, and predefined endpoints.
- The literature recommends larger, randomized studies with clear endpoints (OS/PFS/response/quality of life), and these will be the project's endpoints.
- Before administration, the patient must be examined by their primary care physician to certify their clinical status. Blood tests must be performed to assess liver and kidney function, the presence of previous inflammation, the degree of cellular redox activity, the residual capacity to reduce free radicals, and the immunological status of the patient participating in the study.
- The volunteer patient must not incur any costs and must sign the informed consent form in advance, stating that he or she is participating in the randomized study and clearly specifying that the study is not for therapeutic purposes but rather for observing the effects of the administration of the preparation for the pathology.
- The infusions must take place exclusively in practices or clinics approved as infusion centers pursuant to the Presidential Decree of January 14, 1997, as amended by the consolidated law on local health (TULS). This applies especially to private medical centers participating, after proof has been provided that the clinic where the infusions are performed complies with the legal requirements.
Data collection and processing
This phase is crucial for evaluating the treatment’s efficacy and calculating the planned endpoints, as well as being the gateway to the Phase 2 clinical trial. For this reason, the analysis of the collected data must be performed at a university medical statistics center and must report both the benefits and any adverse effects noted in the short and medium term (the patient remains under periodic observation for 6 months), the success and failure rates, and the calculation of the risk/benefit ratio.
Ethical approval
The study protocol will be conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice (GCP) guidelines. Ethical approval will be obtained from the appropriate Institutional Ethics Committee before initiation of patient recruitment.
Informed consent
Written informed consent will be obtained from all participants prior to enrollment in the study. Participants will be informed regarding the objectives of the study, potential risks, expected benefits, confidentiality measures, and their right to withdraw from the study at any time without affecting their standard medical care.
- Shah T, Krishnamachary B, Wildes F, Wijnen JP, Glunde K, Bhujwalla ZM. Molecular causes of elevated phosphoethanolamine in breast and pancreatic cancer cells. Cancer Res. 2018. Available from: https://doi.org/10.1002/nbm.3936
- Glunde K. Phosphoethanolamine accumulation protects cancer cells under glutamine starvation through downregulation of PCYT2. Cell Rep. 2019. Available from: https://doi.org/10.1016/j.celrep.2019.08.087
- Vance DE. Phospholipid metabolism and cancer. 2014.
- Stoica C, Ferreira AK, Hannan K, Bakovic M. Bilayer forming phospholipids as targets for cancer therapy. Int J Mol Sci, . 2022 May 9;23(9):5266. Available from: https://doi.org/10.3390/ijms23095266
- Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44-84. Available from: https://doi.org/10.1016/j.biocel.2006.07.001
- Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020. Available from: https://doi.org/10.1016/j.ccell.2020.06.001
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. Available from: https://doi.org/10.1016/j.cell.2011.02.013
- Piazza R. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood. 2015;125(3):499‑509. Available from: https://doi.org/10.1182/blood-2014-06-579466
- Fontana D, Gambacorti-Passerini C, Piazza R. Impact of ETNK1 somatic mutations on phosphoethanolamine synthesis, ROS production and DNA damage. Mol Cell Oncol, 2021 Feb 19;8(2):1877598. Available from: https://doi.org/10.1080/23723556.2021.1877598
- Shuai W, Zuo Z, Li N, Garces S, Jelloul FZ, Ok CY, et al. ETNK1 mutation occurs in a wide spectrum of myeloid neoplasms and is not specific for atypical chronic myeloid leukemia. Cancer. 2023;129(6):878‑889. Available from: https://doi.org/10.1002/cncr.34616
- Tessier S, Ali NA, Trinh M, Brunner A, Fontana D, Gambacorti-Passerini C, et al. ETNK1 mutations define a distinct subset of myeloid neoplasms with unique cytogenetic and molecular features. Blood Suppl. 2025/2026.Available from: https://doi.org/10.1182/blood-2025-3864