
More Information
Submitted: October 17, 2023 | Approved: October 25, 2023 | Published: October 26, 2023
How to cite this article: Sitovskaya D, Krapivin M, Sokolova T, Zabrodskaya Y. Diffuse Pediatric-Type High-Grade Glioma H3-/IDH-wildtype with MYCN Deletion and Constitutional Mismatch Repair Deficiency: Case Presentation Arch Case Rep. 2023; 7: 053-057.
DOI: 10.29328/journal.acr.1001079
Copyright License: © 2023 Sitovskaya D, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Pediatric-type high-grade glioma; MYCN deletion; Mismatch repair deficiency

Diffuse Pediatric-Type High-Grade Glioma H3-/IDH-wildtype with MYCN Deletion and Constitutional Mismatch Repair Deficiency: Description of the case Presentation
Darya Sitovskaya1,2* , Mikhail Krapivin3, Tatyana Sokolova1 and Yulia Zabrodskaya1,4
, Mikhail Krapivin3, Tatyana Sokolova1 and Yulia Zabrodskaya1,4
1Polenov Neurosurgical Institute, Branch of Almazov National Medical Research
Centre, 197341 St. Petersburg, Russia
2Department of Pathology with a course in forensic medicine named after D.D.
Lochov, St. Petersburg State Pediatric Medical University, 194100 St. Petersburg, Russia
3Almazov National Medical Research Centre, 197341 St. Petersburg, Russia
4Department of Pathology, Mechnikov North-West State Medical University, 191015 St.
Petersburg, Russia
*Address for Correspondence: Darya Sitovskaya, Polenov Neurosurgical Institute, Branch of Almazov National Medical Research Centre, 197341 St. Petersburg, Russia, Email: [email protected]
Diffuse pediatric-type high-grade glioma H3-wildtype and IDH-wildtype (pHGG H3/IDH WT) is a heterogeneous entity that is currently defined by a combination of highly malignant morphology, glial or primitive neuroectodermal differentiation, and a number of molecular features. Depending on the DNA methylation profile in pHGG H3/IDH WT, three molecular subgroups are distinguished, one of which (pHGG MYCN) is characterized by amplification of the indicated gene. We report a unique case of pHGG H3/IDH WT in a 19-year-old girl with a deletion of the MYCN gene and constitutional mismatch repair deficiency syndrome.
Diffuse pediatric-type high-grade glioma H3-wildtype and IDH-wildtype (pHGG H3/IDH WT) is a heterogeneous entity that is currently defined by a combination of highly malignant morphology, glial or primitive neuroectodermal differentiation, and has a number of molecular features. Initial testing should exclude changes in histone H3 and the IDH1 or IDH2 genes (isocitrate dehydrogenase 1/2) [1]. Alterations commonly found in these tumors include PDGFRA (platelet-derived growth factor receptor, alpha) amplification or mutation, TP53 (tumor protein p53) mutation, NF1 (neurofibromin 1) alterations, EGFR (epidermal growth factor receptor) amplification or mutation, or MYCN amplification (myelocytomatosis, neuroblastoma-derived). Depending on the DNA methylation profile in pHGG H3/IDH WT, three molecular subgroups are distinguished: pHGG RTK1 (receptor tyrosine kinase), pHGG RTK2 and pHGG MYCN. pHGG RTK1 is enriched for PDGFRA amplifications (~33% of cases), pHGG RTK2 is enriched for EGFR amplifications (~50% of cases), and TERT (telomerase reverse transcriptase) promoter mutations (~64% of cases) [2] and pHGG MYCN is enriched for MYCN amplifications (~50% of cases).
pHGG H3/IDH WT represents a large proportion (40%) of pediatric high-grade gliomas, with pHGG MYCN and pHGG RTK2 representing the largest and smallest subgroups, respectively. Regarding etiology, it is noteworthy that the pHGG RTK1 subgroup includes most radiation-induced gliomas developing in patients previously treated for medulloblastoma or acute lymphoblastic leukemia, as well as most gliomas arising in syndromic contexts (eg, Li Fraumeni, constitutional deficiency mismatch repair and Lynch syndrome) [3-5].
Except for a higher degree of genomic instability associated with radiation-induced DNA damage, no significant biological differences have been identified between sporadic and radiation-induced pHGG RTK1 [4,5]. In terms of localization, the vast majority of pHGG H3/IDH WT occurs in the supratentorial anatomical compartment. A minority of pHGG MYCN (about 15%) originate in the brainstem [6-8]. Regarding outcome, the overall prognosis of pHGG H3/IDH WT is poor (WHO grade 4). pHGG MYCN is associated with the lowest survival rates [2], and in this subgroup pontine tumors behave more aggressively than supratentorial counterparts, likely due to tumor location, with median overall survival of 16.5 and 1.5 months for supratentorial and HGGMYCN bridges, respectively [9]. The occurrence of TP53, ATRX (alpha-thalassemia receptor, X-linked), and mismatch repair gene mutations in the context of pHGG H3/IDH WT have been associated with adverse outcomes [10].
However, cases of MYCN deletion in pHGG H3/IDH WT have not previously been reported.
Patient K., 19 years old, was admitted to the Polenov Neurosurgical Institute – Branch of Almazov National Medical Research Center in St. Petersburg, Russia. It is known that a year before hospitalization, she began to experience diffuse headaches, dizziness, and episodes of “darkening” in her eyes. She was treated with drug therapy, which did not bring any effect. After 7 months, the headaches intensified and became localized in the left temporal and parietal region, and nausea appeared. At the height of pain, mainly in the morning, vomiting occurred. The neurologist recommended performing magnetic resonance imaging (MRI) of the brain. According to the results of the MRI, an intracerebral multinodular solid formation of a heterogeneous structure with inclusions of multiple small cysts was visualized in the structure of the left frontal, temporal, and occipital lobes. The tumor dimensions were up to 84×45×70 mm (Figure 1). Signs of mild perifocal edema and mass effect were noted. The median structures were shifted to the right by up to 7 mm. The patient underwent surgical treatment including subtotal tumor removal with Awake under neurolinguistic control and ultrasound navigation.
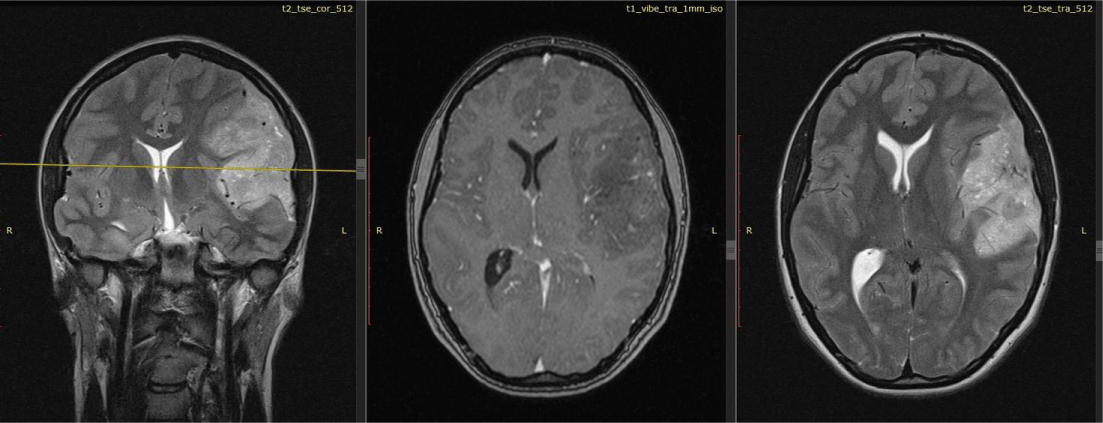
Figure 1: MRI results. Multinodular tumor of the left frontal, temporal, occipital lobes measuring 84×45×70 mm.
Histological examination of the surgical material revealed a malignant glial tumor of a diffuse structure (Figure 2A). The tumor had high cellularity, consisting of cells with rounded hyperchromatic polymorphic nuclei, which were located on a loose fibrillar background. Cellular nuclear polymorphism was moderately expressed, with a few bi- and multinucleated cells, and foci consisting of small monomorphic “homonuclear” cells with rounded hyperchromic nuclei being detected. In the stroma of the tumor, vascular cavities of different sizes were found, some with the proliferation of the endothelium and the formation of glomeruli; thin-walled vessels of small caliber were also found, partially branching like a “chain-link mesh”; signs of hyalinosis of the walls and plethora, and focal hemorrhages were present. Cysts filled with weakly basophilic contents, and foci of calcification deposits were also observed. Few mitoses and apoptotic bodies were present, but no necrosis was detected. Invasion into the cortex and spread through the molecular layer were observed. When immunohistochemical (IHC) examination was performed, the cytoplasm of tumor cells diffusely expressed GFAP+ (Figure 2B). There was a high proliferative activity of Ki67/MIB1 (10-12%, with foci up to 15%; Figure 2C), expression of IDH-1 (Figure 2D), and no detection of H3K27M. Additionally, microsatellite instability was detected with a complete loss of expression of the mismatch repair marker MSH6 by tumor cells (Figure 2E). Antibodies from Dako (Denmark) and an EnVision imaging system were used. Histological analysis and microphotography were performed using a Leica DM2500 M microscope equipped with a DFC320 digital camera and an IM50 image manager (Leica Microsystems, Wetzlar, Germany).
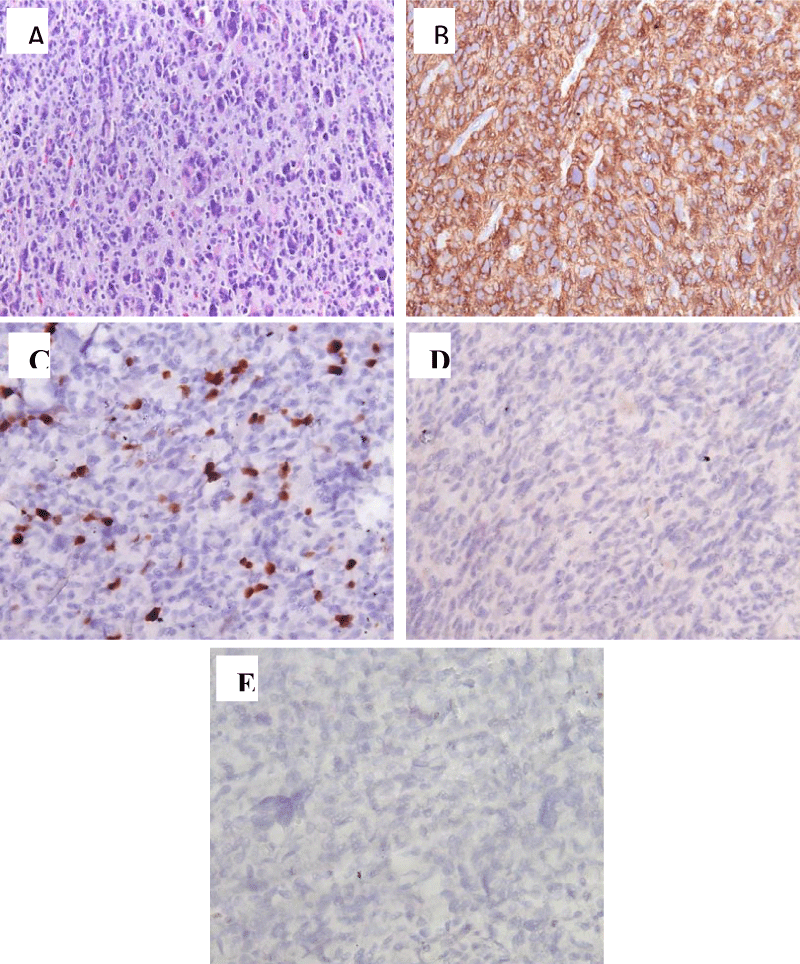
Figure 2: Results of histological and immunohistochemical examination. A: Highly malignant glial tumor infiltrating the cortex. H&E stain, ×200. B: IHC with antibodies to GFAP, ×200; C: Proliferative activity according to Ki67 10% - 15%, ×400; D: IHC with antibodies to IDH 1r132h, ×200. E: IHC with antibodies to MSH6, ×400.
The diagnosis of molecular pathology revealed that the polymerase chain reaction (PCR) method did not detect mutations in the IDH1 and IDH2 genes, the L858R mutation EGFR gene, or deletions. Direct Sanger sequencing also did not detect mutations in the H3F3A (H3 histone, family 3a) gene. As an oligodendroglia-like component was identified in the tumor structure, fluorescent in situ hybridization (FISH) was performed using fluorescent probes, which revealed no rearrangements affecting CHD5 (chromodomain helicase DNA-binding protein 5, 1p36) (Cytotest) и GLTSCR1 (glioma tumor suppressor candidate region gene 1, 19q13.33). To determine the copy number of the MYCN gene, fluorescent in situ hybridization was performed using commercial fluorescent probes (MYCN (2p24) / AFF3 (alf transcription elongation factor 3, 2q11), Kreatech FISH probes, Leica Biosystems, USA, KI-10706). The red signals of the probes were specific for the MYCN gene, and the green signals were specific for AFF3, which acted as a cell ploidy control. This DNA probe system was used to determine MYCN gene amplification in aggressive tumors. In the tissue sample under study, there were 5 fragments, in 4 of which most of the cells had fewer red signals than green ones (Figure 3), indicating a deletion of the region of the short arm of chromosome 2 containing the MYCN gene (region 2p24). Research result: nuc ish(MYCNх2,AFF3х1)[3/188]/(MYCNх2~3,AFF3х3~4)[21/188]/(MYCN,AFF3)x2[75/188]/(MYCNх1,AFF3х2)[89/188].
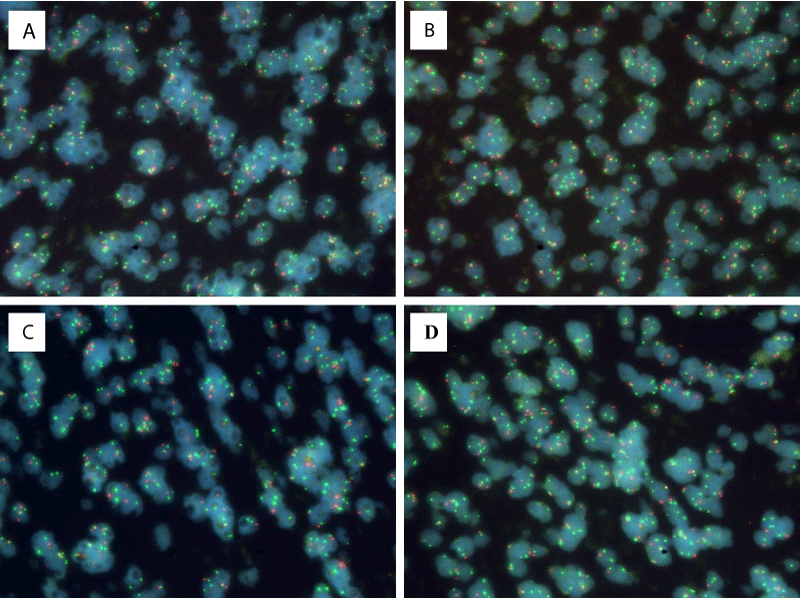
Figure 3: FISH results (A-D): Most of the cells have fewer red signals than green ones (ploidy control), which indicates a deletion of the region of the short arm of chromosome 2 containing the MYCN gene (region 2p24), ×630.
Currently, the patient is still alive, having undergone 28 courses of chemotherapy with temozolomide and radiation therapy with a total dose of 60 Gray. There were no signs of disease progression or tumors in other locations.
MYCN (OMIM 164840), the human gene encoding N-Myc, was first identified as an oncogene amplified in human neuroblastoma [11], a tumor characterized by the presence of undifferentiated neuroblasts [12]. The level of MYCN expression correlates with the prognosis of neuroblastoma [13] and overexpression of the human MYCN gene causes neuroblastoma in mice [14]. MYCN overexpression is also observed in other nervous system tumors such as medulloblastoma, glioblastoma, retinoblastoma, and spinal ependymoma, as well as in tumors outside the nervous system, such as neuroendocrine prostate cancer, nephroblastoma (Wilms tumor), and many others [15-17]. The nucleotide sequence of MYCN is very similar to that of the oncogene MYC [18]. Additionally, in addition to tumorigenesis in the nervous system, the MYCN oncogene has been shown to play a critical role in neurogenesis and oligodendrogenesis in the healthy adult brain [19]. However, the prognosis of tumors with MYCN gene deletion still needs to be studied.
Constitutional mismatch repair deficiency (CMMRD) is an autosomal recessive disorder caused by biallelic variants in one of four mismatch repair genes: MLH1 (DNA mismatch repair protein 1), MSH2 (MutS homolog 2), MSH6 (MutS homolog 6), and PMS2 (postmeiotic segregation increased, s. cerevisiae, 2). This condition can cause a wide range of tumors in childhood, adolescence, and young adulthood, with high-grade gliomas being the most common type. In approximately 60% of cases, CMMRD is caused by biallelic pathogenic variants of PMS2, in 20-30% by biallelic pathogenic variants of MSH6, and in 10-20% by biallelic pathogenic variants of MLH1 or MSH2 [20,21].
The literature describes isolated cases of deletion of the short arm of chromosome 2 at the organismal level, associated with multiple congenital malformations [22,23]. Therefore, further research is required to analyze genetic variants in malignant brain tumors.
Thus, a 19-year-old patient with constitutional mismatch repair deficiency syndrome developed a highly malignant diffuse pediatric-type glioma H3-wildtype and IDH-wildtype, in which a unique defect was discovered: deletion of the MYCN gene (region 2p24). The effect on the course of the disease and prognosis requires further study.
Institutional review board statement
The work was carried out according to the principles of voluntariness and confidentiality in accordance with Federal Law “On the Basics of Health Protection of Citizens in Russian Federation” 21.11.2011 N 323-FZ, and the Helsinki Declaration on Human Rights. The patient signed informed voluntary consent for the study.
- WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France): International Agency for Research on Cancer; 2021. (WHO classification of tumours series, 5th ed.; vol. 6). https://publications.iarc.fr/601.
- Korshunov A, Schrimpf D, Ryzhova M, Sturm D, Chavez L, Hovestadt V, Sharma T, Habel A, Burford A, Jones C, Zheludkova O, Kumirova E, Kramm CM, Golanov A, Capper D, von Deimling A, Pfister SM, Jones DTW. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017 Sep;134(3):507-516. doi: 10.1007/s00401-017-1710-1. Epub 2017 Apr 11. PMID: 28401334.
- López GY, Van Ziffle J, Onodera C, Grenert JP, Yeh I, Bastian BC, Clarke J, Oberheim Bush NA, Taylor J, Chang S, Butowski N, Banerjee A, Mueller S, Kline C, Torkildson J, Samuel D, Siongco A, Raffel C, Gupta N, Kunwar S, Mummaneni P, Aghi M, Theodosopoulos P, Berger M, Phillips JJ, Pekmezci M, Tihan T, Bollen AW, Perry A, Solomon DA. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol. 2019 Jan;137(1):139-150. doi: 10.1007/s00401-018-1906-z. Epub 2018 Sep 8. PMID: 30196423; PMCID: PMC6589431.
- DeSisto J, Lucas JT Jr, Xu K, Donson A, Lin T, Sanford B, Wu G, Tran QT, Hedges D, Hsu CY, Armstrong GT, Arnold M, Bhatia S, Flannery P, Lemma R, Hardie L, Schüller U, Venkataraman S, Hoffman LM, Dorris K, Mulcahy Levy JM, Hankinson TC, Handler M, Liu AK, Foreman N, Vibhakar R, Jones K, Allen S, Zhang J, Baker SJ, Merchant TE, Orr BA, Green AL. Comprehensive molecular characterization of pediatric radiation-induced high-grade glioma. Nat Commun. 2021 Sep 20;12(1):5531. doi: 10.1038/s41467-021-25709-x. PMID: 34545084; PMCID: PMC8452624.
- Deng MY, Sturm D, Pfaff E, Sill M, Stichel D, Balasubramanian GP, Tippelt S, Kramm C, Donson AM, Green AL, Jones C, Schittenhelm J, Ebinger M, Schuhmann MU, Jones BC, van Tilburg CM, Wittmann A, Golanov A, Ryzhova M, Ecker J, Milde T, Witt O, Sahm F, Reuss D, Sumerauer D, Zamecnik J, Korshunov A, von Deimling A, Pfister SM, Jones DTW. Radiation-induced gliomas represent H3-/IDH-wild type pediatric gliomas with recurrent PDGFRA amplification and loss of CDKN2A/B. Nat Commun. 2021 Sep 20;12(1):5530. doi: 10.1038/s41467-021-25708-y. PMID: 34545083; PMCID: PMC8452680.
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, Zagzag D, Foreman NK, Donson A, Hegert JV, Smith A, Chan J, Lafay-Cousin L, Dunn S, Hukin J, Dunham C, Scheinemann K, Michaud J, Zelcer S, Ramsay D, Cain J, Brennan C, Souweidane MM, Jones C, Allis CD, Brudno M, Becher O, Hawkins C. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014 May;46(5):451-6. doi: 10.1038/ng.2936. Epub 2014 Apr 6. PMID: 24705254; PMCID: PMC3997489.
- Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA, Zheludkova O, Milde T, Witt O, Kulozik AE, Reifenberger G, Jabado N, Perry A, Lichter P, von Deimling A, Pfister SM, Jones DT. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015 May;129(5):669-78. doi: 10.1007/s00401-015-1405-4. Epub 2015 Mar 10. PMID: 25752754.
- Tauziède-Espariat A, Debily MA, Castel D, Grill J, Puget S, Sabel M, Blomgren K, Gareton A, Dangouloff-Ros V, Lechapt E, Boddaert N, Varlet P. An integrative radiological, histopathological and molecular analysis of pediatric pontine histone-wildtype glioma with MYCN amplification (HGG-MYCN). Acta Neuropathol Commun. 2019 Jun 10;7(1):87. doi: 10.1186/s40478-019-0738-y. Erratum in: Acta Neuropathol Commun. 2019 Aug 14;7(1):131. PMID: 31177990; PMCID: PMC6556947.
- Tauziède-Espariat A, Debily MA, Castel D, Grill J, Puget S, Roux A, Saffroy R, Pagès M, Gareton A, Chrétien F, Lechapt E, Dangouloff-Ros V, Boddaert N, Varlet P. The pediatric supratentorial MYCN-amplified high-grade gliomas methylation class presents the same radiological, histopathological and molecular features as their pontine counterparts. Acta Neuropathol Commun. 2020 Jul 9;8(1):104. doi: 10.1186/s40478-020-00974-x. PMID: 32646492; PMCID: PMC7346460.
- Hong L, Shi ZF, Li KK, Wang WW, Yang RR, Kwan JS, Chen H, Li FC, Liu XZ, Chan DT, Li WC, Zhang ZY, Mao Y, Ng HK. Molecular landscape of pediatric type IDH wildtype, H3 wildtype hemispheric glioblastomas. Lab Invest. 2022 Jul;102(7):731-740. doi: 10.1038/s41374-022-00769-9. Epub 2022 Mar 24. PMID: 35332262.
- Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983 Sep 15-21;305(5931):245-8. doi: 10.1038/305245a0. PMID: 6888561.
- Wright JH. NEUROCYTOMA OR NEUROBLASTOMA, A KIND OF TUMOR NOT GENERALLY RECOGNIZED. J Exp Med. 1910 Jul 23;12(4):556-61. doi: 10.1084/jem.12.4.556. PMID: 19867342; PMCID: PMC2124805.
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984 Jun 8;224(4653):1121-4. doi: 10.1126/science.6719137. PMID: 6719137.
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997 Jun 2;16(11):2985-95. doi: 10.1093/emboj/16.11.2985. PMID: 9214616; PMCID: PMC1169917.
- Ruiz-Pérez MV, Henley AB, Arsenian-Henriksson M. The MYCN Protein in Health and Disease. Genes (Basel). 2017 Mar 30;8(4):113. doi: 10.3390/genes8040113. PMID: 28358317; PMCID: PMC5406860.
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 Oct 23;455(7216):1061-8. doi: 10.1038/nature07385. Epub 2008 Sep 4. Erratum in: Nature. 2013 Feb 28;494(7438):506. PMID: 18772890; PMCID: PMC2671642.
- Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, Scheie D, Kruse A, Harter PN, Kastelan M, Wagner M, Hartmann C, Benzel J, Maass KK, Khasraw M, Sträter R, Thomas C, Paulus W, Kratz CP, Witt H, Kawauchi D, Herold-Mende C, Sahm F, Brandner S, Kool M, Jones DTW, von Deimling A, Pfister SM, Reuss DE, Pajtler KW. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019 Dec;138(6):1075-1089. doi: 10.1007/s00401-019-02056-2. Epub 2019 Aug 14. PMID: 31414211; PMCID: PMC6851394.
- Kohl NE, Legouy E, DePinho RA, Nisen PD, Smith RK, Gee CE, Alt FW. Human N-myc is closely related in organization and nucleotide sequence to c-myc. Nature. 1986 Jan 2-8;319(6048):73-7. doi: 10.1038/319073a0. PMID: 3510398.
- Chen J, Guan Z. Function of Oncogene Mycn in Adult Neurogenesis and Oligodendrogenesis. Mol Neurobiol. 2022 Jan;59(1):77-92. doi: 10.1007/s12035-021-02584-7. Epub 2021 Oct 8. PMID: 34625907; PMCID: PMC8786763.
- Durno C, Ercan AB, Bianchi V, Edwards M, Aronson M, Galati M, Atenafu EG, Abebe-Campino G, Al-Battashi A, Alharbi M, Azad VF, Baris HN, Basel D, Bedgood R, Bendel A, Ben-Shachar S, Blumenthal DT, Blundell M, Bornhorst M, Bronsema A, Cairney E, Rhode S, Caspi S, Chamdin A, Chiaravalli S, Constantini S, Crooks B, Das A, Dvir R, Farah R, Foulkes WD, Frenkel Z, Gallinger B, Gardner S, Gass D, Ghalibafian M, Gilpin C, Goldberg Y, Goudie C, Hamid SA, Hampel H, Hansford JR, Harlos C, Hijiya N, Hsu S, Kamihara J, Kebudi R, Knipstein J, Koschmann C, Kratz C, Larouche V, Lassaletta A, Lindhorst S, Ling SC, Link MP, Loret De Mola R, Luiten R, Lurye M, Maciaszek JL, MagimairajanIssai V, Maher OM, Massimino M, McGee RB, Mushtaq N, Mason G, Newmark M, Nicholas G, Nichols KE, Nicolaides T, Opocher E, Osborn M, Oshrine B, Pearlman R, Pettee D, Rapp J, Rashid M, Reddy A, Reichman L, Remke M, Robbins G, Roy S, Sabel M, Samuel D, Scheers I, Schneider KW, Sen S, Stearns D, Sumerauer D, Swallow C, Taylor L, Thomas G, Toledano H, Tomboc P, Van Damme A, Winer I, Yalon M, Yen LY, Zapotocky M, Zelcer S, Ziegler DS, Zimmermann S, Hawkins C, Malkin D, Bouffet E, Villani A, Tabori U. Survival Benefit for Individuals With Constitutional Mismatch Repair Deficiency Undergoing Surveillance. J Clin Oncol. 2021 Sep 1;39(25):2779-2790. doi: 10.1200/JCO.20.02636. Epub 2021 May 4. PMID: 33945292; PMCID: PMC8407605.
- Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, Gerdes AM, Goldberg Y, Ilencikova D, Muleris M, Duval A, Lavoine N, Ruiz-Ponte C, Slavc I, Burkhardt B, Brugieres L; EU-Consortium Care for CMMRD (C4CMMRD). Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet. 2014 Jun;51(6):355-65. doi: 10.1136/jmedgenet-2014-102284. Epub 2014 Apr 15. PMID: 24737826.
- Saal HM, King LJ, Zimmerman D, Johnson RC, Carr AG, Samango-Sprouse CA, Stanley W. Loss of the N-myc oncogene in a patient with a small interstitial deletion of the short arm of chromosome 2. Am J Med Genet. 1996 Dec 30;66(4):373-7. doi: 10.1002/(SICI)1096-8628(19961230)66:4<373::AID-AJMG1>3.0.CO;2-M. PMID: 8989454.
- Neidich J, Zackai E, Aronson M, Emanuel BS. Deletion of 2p: a cytogenetic and clinical update. Am J Med Genet. 1987 Jul;27(3):707-10. doi: 10.1002/ajmg.1320270326. PMID: 3477100.