
More Information
Submitted: 03 March 2020 | Approved: 17 March 2020 | Published: 18 March 2020
How to cite this article: Solomou A, Patriarcheas V, Kraniotis P, Eliades A. Epstein-Barr infection causing toxic epidermal necrolysis, hemophagocytic lymphohistiocytosis and cerebritis in a pediatric patient. Arch Case Rep. 2020; 4: 015-019.
DOI: 10.29328/journal.acr.1001032
Copyright License: © 2020 URAKÇI Z, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Epstein-Barr infection causing toxic epidermal necrolysis, hemophagocytic lymphohistiocytosis and cerebritis in a pediatric patient
Aikaterini Solomou1*, Vasileios Patriarcheas1, Pantelis Kraniotis1 and Andreas Eliades2
1Department of Radiology, MRI Unit, University Hospital of Patras, Greece
2Department of Pediatrics, Intensive Care Unit, University Hospital of Patras,
Greece
*Address for Correspondence: Aikaterini Solomou, Department of Radiology, MRI Unit, University Hospital of Patras, Greece, Tel: +302610999422; Email: [email protected]
Toxic epidermal necrolysis -the most serious variant of Steven Johnson Syndrome -arises as the result of cell-mediated cytotoxic reaction against keratinocytes. Most common inciting factors include drugs, and infections. On the other hand, Hemophagocytic lymphohistiocytosis (HLH), is a syndrome characterized by enormous immune response in the absence of down-regulation of activated immune cells resulting in cytokine storm causing severe tissue damage.
Up to date, several cases of concomitance of Toxic Epidermal Necrolysis (TEN) and Hemophagocytic Lympohystiocytosis (HLH) in pediatric patients have been reported. Both situations can be fatal and pediatricians should be aware that these two clinical entities are not mutually exclusive, to the contrary they may coexist.
We herein describe a case of Toxic Epidermal Necrolysis, complicated with Hemophagocytic Lymphohistiocytosis with Central Nervous System involvement due to EBV infection.
Toxic epidermal necrolysis (TEN) is a life-threatening, immunological reaction, characterized by severe mucocutaneous destruction with detachment of the epidermis and mucous membranes. TEN is the most severe variant of Steven Johnson Syndrome (SJS) affecting more than 30% of body surface area (BSA) [1].
Although the precise pathophysiological mechanism of SJS/TEN is not completely understood, it is known that there is a cell-mediated cytotoxic reaction against keratinocytes resulting in apoptosis [2]. Drugs such as antibacterial sulfonamides, allopurinol, anticonvulsants and NSAIDS, constitute the commonest trigger of SJS/TEN [3]. However some cases of SJS/TEN are not drug-induced. Among non drug factors, bacterial (M. Pneumoniae, Group A Beta Streptococci) and viral infections (EBV, CMV, HSV, HHV-6&7, Parvovirus) are the second most common triggering events [4,5]. Rarely SJS/TEN can result in the setting of malignancy, while even rarer; it can be a manifestation of acute graft-versus-host disease after bone marrow transplantation (BMT) [6].
Clinical manifestations of SJS/TEN include fever (often high grade), malaise, and mucocutaneous lesions. Most patients, experience prodromal flu-like symptoms, ocular pruritus and dysphagia which last 1-7 days [3]. The skin lesions typically begin with purpuric macules on the face and upper torso. They are symmetrically distributed and over time become larger and merge. As the disease progresses, vesicles and blisters form and within days the skin begins to slough. Clinical examination reveals positive Nikolsky sign (i.e. detachment of epidermis from dermis after gentle pressure) [7]. Mucosal involvement is present in the majority of cases and it can follow or precede skin lesions [8]. SJS/TEN has a high mortality rate (> 30%) due to the fact that epidermal lesions make patients prone to insensible fluid loss, electrolyte imbalance and thermoregulatory dysfunction. For this reason, early diagnosis, withdrawal from the inciting agent and supportive care, are prerequisite [7]. Unfortunately, skin lesion biopsy is only supportive -neither specific, nor diagnostic-, which implies that SJS/TEN diagnosis, is mainly clinical. In addition to the above, another limitation is that there are not universally accepted criteria making diagnosis apart from crucial, challenging [3].
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome characterized by enormous immune response in the absence of down-regulation of activated immune cells [9]. Natural killer cells, as well as Cytotoxic T Lymphocytes, which are the responsible cells for the homeostatic down regulation of the immune response, fail to eliminate activated macrophages [10]. Thus, this lack of negative feedback leads to hyperactivated macrophages that secrete excessive amounts of cytokines, resulting in a cytokine storm. This severe hyperinflammation state ultimately causes severe tissue damage that can lead to multisystem organ dysfunction and failure [11].
HLH is classified as primary (i.e. familial) or secondary. Primary HLH is an autosomal recessive disorder, caused by mutations in genes involved in the secretory lysosome-dependent exocytosis pathway. It has an estimated incidence of around 1:50.000 births and is considered a fatal disease with a median survival of less than 2 months if untreated. Familial HLH is further sub-classified into 5 types according to the gene involved. Secondary HLH is developed as a result of severe immune response into stimuli such as infections, malignancies and rheumatologic disorders. It has been described in both immunocompromised and immunocompetent patients, with the majority of patients not being immunosuppressed [10,11].
The typical clinical presentation involves a prolonged high grade fever in combination with splenomegaly and cytopenias. In the majority of cases, the clinical features are similar regardless of whether HLH is primary or secondary.
Common findings include, liver dysfunction and central nervous system symptoms ranging from seizures to encephalopathy while less common findings include lympa-denopathy, jaundice and rash [11].
Despite the fact that the above signs and symptoms can occur in a variety of clinical entities, their combination is strongly suggestive of HLH. For this purpose, the Histiocyte Society presented the first set of diagnostic and therapeutic guidelines in 1991 and in 1994 respectively. Diagnostic guidelines were updated in 2004, according to the HLH-2004 trial [12]. Fulfillment of five of the eight criteria set the diagnosis. In patients who underwent molecular diagnosis consistent with HLH, none of the rest of the diagnostic criteria is required.
A 3.5-year-old girl who was hospitalized for fever with altered level of consciousness due to EBV infection at the pediatrics clinic, was referred to the pediatric intensive care unit due to spreading erythymatous rash affecting > 90% of the body surface with positive Nikolsky sign (Figure 1).
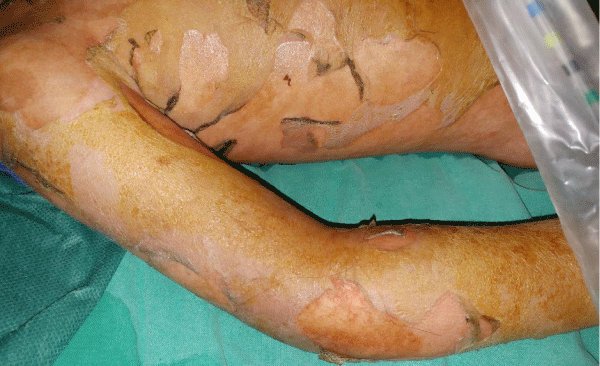
Figure 1: Picture of the torso and the right upper limb of the patient showing the severe and extensive skin changes.
She also exhibited oral mucositis, spleen and liver enlargement. Antibiotics and fluid resuscitation therapy were administered.
Because of the severity of the illness, she required ventilatory support. A nasogastric tube (NGT), was placed for nutritional support.
Laboratory investigation revealed a decrease in white blood cells (2000/μl) and platelets (95.000/μl), elevated serum ferritin (6600 μg/L) and hypertriglyceridemia (400 mg/dL).
Due to the high suspicion of HLH, a bone marrow biopsy performed that showed hemophagocytosis. Subsequently, our patient was treated according to the HLH protocol with Etoposide, Dexamethazone and Cyclosporine. Polymerase Chain Reaction (PCR) of the bone marrow sample, detect Epstein-Barr virus DNA (3.3 x 103 copies/ml).
Moreover, due to cytopenias our patient received Granulocyte Colony Stimulating Factor (GCSF) growth factor in parallel with wide spectrum antibiotics and antifungal therapy (collistin, tazocin, meropenem, vancomycin, collistin, tazocin, micafungin).
The patient was discharged from mechanical ventilation during the 10th day of hospitalization. However, during the upcoming days she exhibited gradual deterioration and 14 days later, she was intubated again. During this phase, our patient developed airway obstruction due to Debris. A cricothyrotomy was performed, to secure the airway.
At this time, she experienced 2 cardiac arrest events subsequent to respiratory distress that were treated successfully. An external pacemaker was installed for 48h.
Bronchoscopy was performed, and revealed large areas of ulceration due to Toxic Epidermal Necrolysis. A second bronchoscopy performed 1week later was normal. Our patient, was discharged from mechanical ventilation during the 28th day and she remained in noninvasive ventilator support [OHIO System].
After extubation, the patient exhibited high blood pressure presumably of central etiology and she received, combined antihypertensive treatment with ACE inhibitor, b-blocker and calcium channel blocker.
During the disease progression, the patient developed severe encephalitis (inability to respond to environmental stimuli and communicate verbally). Because of severe cutaneous lesions, lumbar puncture could not be performed and for this reason MR imaging was the only way to evaluate CNS involvement.
The patient underwent brain MRI which did not reveal any lesions or diffusion restriction in the brain parenchyma. The temporal horns were within normal limits (Figure 2).
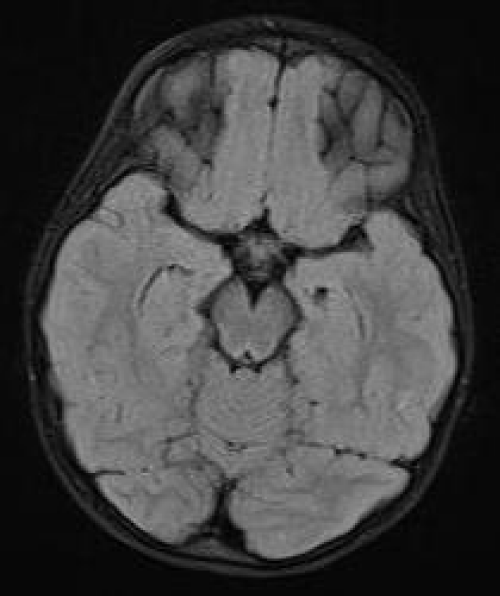
Figure 2: 1st MRI. Axial FLAIR. The temporal horns are within normal limits. There is no abnormal signal in the brain parenchyma.
A new MRI one month later showed lesions centered in the basal ganglia with high signal intensity on FLAIR (Figure 3a) and low signal intensity on T1-weighted images. There was also true restriction on diffusion-weighted images in the external and internal capsules (Figure 3b).
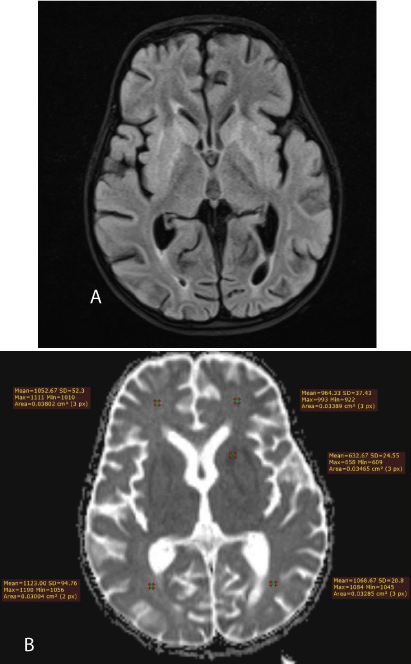
Figure 3: a) 2nd MRI. The high signal intensity mainly affects the internal and external capsules. b) 2nd MRI. Axial ADC map. There is true diffusion restriction only in the external and internal capsules, according the low ADC values, as measured.
The patient was submitted to a new brain MRI, 20 days later, which revealed that the lesions in the basal ganglia were more extensive on the FLAIR (Figure 4a) and DW images. However, the putamen and the globus pallidus bilaterally were hyper intense on the T1-weighted images (Figure 4b). The findings could be compatible with the presence of methemoglobin, due to an underlying toxic insult. Furthermore, the temporal horns were dilated, with diffuse brain atrophy. The corpus callosum was slightly thinned with abnormal signal in its posterior part (Figure 4c).
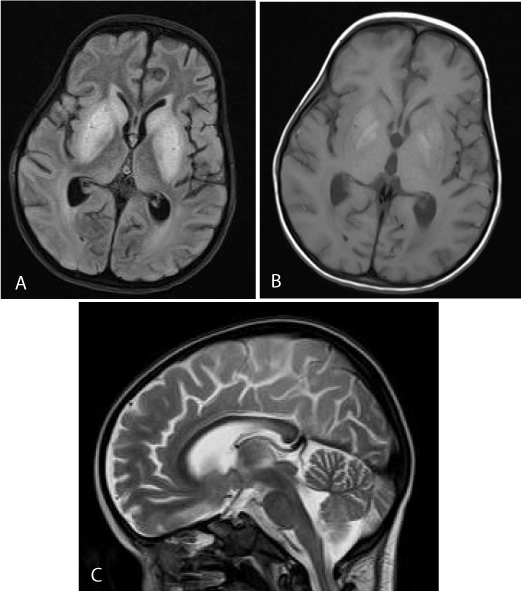
Figure 4: a) 3rd MRI. Axial FLAIR. There is diffuse hyperintensity in the basal ganglia and caudate nuclei. b) 3rd MRI. Axial T1WI. Hyperintensity in the putamen and globus pallidus bilaterally may represent methemoglobin, due to petechial hemorrhage. c) 3rd MRI SAG T2WI. The corpus callosum is slightly thinned with abnormal signal in its posterior part.
On the follow-up MRIs, during the next 2 years, the dilatation of the ventricular system was more prominent and severe with residual gliosis along the basal ganglia (Figure 5). There was no evidence of restriction on the diffusion-weighted images.
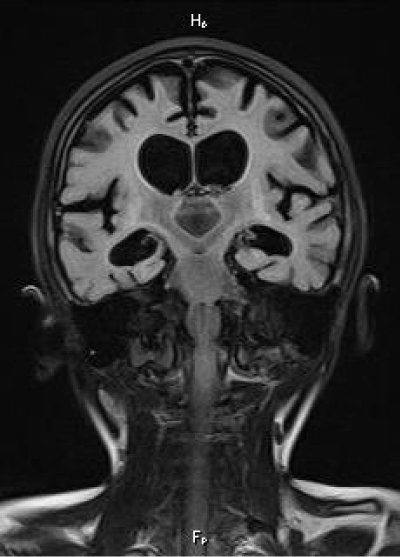
Figure 5: Final, 2 year follow-up MRI. Coronal FLAIR. There is diffuse brain atrophy and ventricular dilatation. Residual gliosis is noted along the basal ganlia.
Notwithstanding the fact that cutaneous maculopapular rashes are described in HLH [13], extensive lesions and widespread epidermal necrosis are extremely rare, and only a few cases of coincidence of SJS/TEN and HLH in pediatric patients have been reported [14-19]. We believe that it is important to highlight that these two life-threatening clinical entities may coexist.
As in the majority of published cases, HLH consequently developed to SJS/TEN in our patient, as well. Both diseases share common triggers, such as drugs and infection. In our case we postulate that EBV infection was the causative factor for TEN as well as for HLH.
We cannot be sure if the appearance of skin lesions prior to HLH is just an observation or if it is indicative of disease progression, as a part of a common pathway of defective activation of CD8+ lymphocytes, where in SJS/TEN the latter attack keratinocytes promoting their apoptosis, while in HLH they fail to suppress macrophage activation.
CNS involvement is not included in the diagnostic criteria; however this is a common manifestation in HLH with an incidence of up to 70% and can occur either at presentation or throughout the course of disease progression. [20].
CNS manifestations occur as the result of brain infiltration by activated lymphocytes and macrophages and exhibit a wide range of symptoms. Long-term deficits (motor or conginitive) are possible to occur. Common manifestations include: altered consciousness seizures, ataxia, meningism, cranial nerve palsy, hemiplegia/tetraplegia and signs of elevated intracranial pressure (ICP). Evaluation of neurologic features requires lumbar puncture (contraindicated in cases with elevated ICP) for CSF analysis and neuroimaging.
MRI is the modality of choice for the evaluation of CNS involvement, especially in patients where CSF analysis cannot be performed, as in our patient, due to skin lesions in the lumbar region.
The symmetrical, bilateral lesions in the basal ganglia, which are hyperintense on T2-weighted and FLAIR images, in addition to diffusion restriction on DWI, are characteristic imaging findings of EBV encephalitis. The diagnosis was in accordance with the positive EBV test and the clinical symptoms of encephalitis with which the little patient presented. There was also involvement of the splenium, the latter being a rare finding of this entity. The diffusion restriction on DWI may be attributed to hemophagocytic infiltration, because of neuronal loss and edema[21].
There was neither involvement of the cortex, nor of the brainstem or the white matter.
Interestingly our patient also showed a progressive cerebral volume loss with dilatation of the ventricular system on the serial brain MRI scans. Extra-axial fluid collections were not observed. Cerebral atrophy was compatible with the HLH. Other imaging findings that may be present with HLH, -but are not specific for the disease-, are high signal intensity white matter, cortical or subcortical lesions on T2-weighted and FLAIR images with or without enhancement. Leptomeningeal enhancement after the administration of gadolinium may be present. In our case, there was no evidence of abnormal enhancement [22]. Referring to TEN/ Steven Johnson Syndrome the imaging findings are not characteristic.
Summarizing we would like to stress that:
ΕBV-HLH is usually first detected as persistent fever that is unresponsive to antibiotics.
In clinical terms it can be classified as a neoplasmatic disease as well as an infectious/reactive disease. In order to establish the diagnosis of EBV-HLH the patient must be EBV positive. On the other hand, rapid diagnosis of HLH is established by a set of diagnostic guidelines (clinical and laboratory findings - Histiocyte Society).
It should be noted that both SJS (Steven Johnson) and HLH have a high mortality rate so concurrence of the two in a patient is especially dangerous and catastrophic. More than 30% of the patients diagnosed with EBV-HLH succumb to the disease or its complications.
Unfortunately the relative literature concerning the coexistence of these clinical entities is very limited. It is however presumed that there may be an abnormal stimulation of lymphocytes, presenting with a double clinical expression. Many points are still undecipherable and the answers may be probably found only at the genetic level. The scope of this case report is to add to the literature and alert the doctors implicated not only for both entities but also for their coexistence, which may bear a heavy toll to the patient.
Study limitation may be the fact that the only evidence provided of a causative implication from initial EBV infection is the detection of EBV DNA in bone marrow biopsied samples. PCR results of DNA, at best, indicates presence per se and do not substantiate activity of the virus in the context of disease phenotype. It is understandable that transcript detection via Real Time – Quantitative PCR approach may not be the standard protocol for laboratory evaluation of patient samples at primary care centres (hospitals). As such, the allegation that EBV infection as a bona fide and sole causative factor should, on the one hand, be taken with caution, but on the other hand still taken into serious consideration. In our opinion the latter should be a strong suggestion, if not a recommendation, for future diagnostic endeavours.
Finally, according to our experience clinicians involved in the diagnosis and management of TEN must be alert to the possibility of coexisting HLH, particularly in cases with severe multi-organ system involvement. As fatal cases of TEN might present symptoms that could ultimately be associated with HLH, treating appropriately could be vital for patient outcome.
In our case the chemotherapeutic treatment was successful
(undetectable EBV DNA copies) and bone marrow transplan-tation was avoided. The patient was discharged from the hospital on day 94 of staying, with severe neurological sequelae. Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal hyperinflammatory condition with both genetic and acquired causes. EBV infection is the most common viral etiologic factor.
Diagnosis of HLH is challenging and this life-threatening condition can be underdiagnosed or misdiagnosed because signs and symptoms of the disease can occur in a variety of clinical entities.
Especially in cases where there is coexistence of TEN/HLH, there is a higher risk for misdiagnosis due to overlapping clinical features.
Physicians should be aware of this rare clinical entity, as of their even more rare coincidence, because initiation of proper treatment is related to more favorable outcome.
Despite the fact that CNS involvement is not included in the diagnostic criteria of HLH, we have to point out that the majority of patients will have CNS manifestations. Hence, a child with unexplained neurologic symptoms and a combination of fever, splenomegaly and cytopenias, should be thoroughly investigated.
- Stern RS, Divito SJ. Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Associations, Outcomes, and Pathobiology-Thirty Years of Progress but Still Much to Be Done. J Invest Dermatol. 2017; 137: 1004–1008. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28411832
- Nassif A1, Bensussan A, Boumsell L, Deniaud A, Moslehi H, et al. Toxic epidermal necrolysis: Effector cells are drug-specific cytotoxic T cells. J Allergy Clin Immunol. 2004; 114: 1209-1215. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15536433
- Ferrandiz-Pulido C1, Garcia-Patos V. A review of causes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Arch Dis Child. 2013; 98: 998-1003. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23873883
- Levi N1, Bastuji-Garin S, Mockenhaupt M, Roujeau JC, Flahault A, et al. Medications as risk factors of Stevens-Johnson syndrome and toxic epidermal necrolysis in children: a pooled analysis. Pediatrics. 2009; 123: 297–304. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19153164
- Kunimi Y, Hirata Y, Aihara M, Yamane Y, Ikezawa Z. Statistical analysis of Stevens-Johnson syndrome caused by Mycoplasma pneumonia infection in Japan. Allergol Int. 2011; 60: 525–532. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22113160
- Macedo FI1, Faris J, Lum LG, Gabali A, Uberti JP, et al. Extensive toxic epidermal necrolysis versus acute graft versus host disease after allogenic hematopoietic stem-cell transplantation: challenges in diagnosis and management. J Burn Care Res. 2014; 35: e431-5. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24476990
- Harris V, Jackson C, Cooper A. Review of Toxic Epidermal Necrolysis. Int J Mol Sci. 2016; 17: 2135. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5187935/
- Revuz J, Roujeau JC, Guillaume JC, Penso D, Touraine R. Treatment of toxic epidermal necrolysis. Créteil's experience. Arch Dermatol. 1987; 123: 1156. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3631999
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: Part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention and treatment. J Am Acad Dermatol. 2013; 69: 187.e1. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23866879
- Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012; 79: 356-361. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22464018
- George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014; 5: 69–86. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4062561/
- Aricò M1, Janka G, Fischer A, Henter JI, Blanche S, et al. Hemophagocytic lymphohis- tiocytosis: Diagnosis, treatment and prognostic factors. Report of 122 children from the international registry. Leukemia 1996; 10: 197-203. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8637226
- Henter JI1, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 48: 124-131. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16937360
- Fardet L1, Galicier L, Vignon-Pennamen MD, Regnier S, Noguera ME, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010; 162: 547–553. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19818067
- Sniderman JD, Cuvelier GD, Veroukis S, Hansen G. Toxic epidermal necrolysis and hemophagocytic lymphohistiocytosis: a case report and literature review. Clin Case Rep. 2015; 3: 121–125. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4352368/
- Fan ZD, Qian XQ, Yu HG. Pancytopenia as an early indicator for Stevens-Johnson syndrome complicated with hemophagocytic lymphohistiocytosis: a case report. BMC Pediatr. 2014; 14: 38. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24512039
- Kawachi Y, Itoh M, Fujisawa Y, Furuta J, Nakamura Y, et al. Epidermal cell necrosis with direct epidermal infiltration of Epstein-Barr virus (EBV)-encoded small nuclear RNA-positive T lymphocytes in a patient with EBV-associated haemophagocytic syndrome. Br J Dermatol. 2007; 157: 1053-1056. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17725675
- Zeng H. Chen X. First case report of Stevens-Johnson syndrome complicated with macrophage activation syndrome. Rheumatol Rep. 2009; 1: 30–31.
- Sharma N, Clark J, Pham H, Efron D, MacGregor D, et al. TEN-like eruption in setting of EBV positive T-cell lymphoproliferative disease with HLH, in a child. Australas. J Dermatol. 2013; 55: e44–e47. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23573996
- Pakran J, Pavithran K, Kuruvila S, Anand M. Coexistence of Stevens-Johnson syndrome and hemophagocytic syndrome. Indian J Pediatr Dermatol. 2013; 14: 83.
- Horne A, Wickström R, Jordan MB, Yeh EA, Naqvi A, et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol. 2017; 19: 3. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5290057/
- Henter J-I, Nennesmo I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. J Pediatrics. 1997; 130: 358–365. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5290057/
- Shieh AC, Guler E, Smith DA, Tirumani SH, Beck RC, et al. Hemophagocytic Lymphohistiocytosis: A primer for Radiologists. AJR Am J Roentgenol. 2020; 214: W11-W19. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31532253