
More Information
Submitted: May 09, 2025 | Approved: May 15, 2025 | Published: May 16, 2025
How to cite this article: Angel-Rodríguez F, Usta-Stavoli J, Cruz-Gonzalez A, Huguet G, Urbina A, Silgado-Guzman D, et al. Synergistic Heterozygosity in Rare Digenic Hereditary Anemias Accurately Diagnosed by whole Exome Sequencing. Arch Case Rep. 2025; 9(5): 176-179. Available from:
https://dx.doi.org/10.29328/journal.acr.1001141
DOI: 10.29328/journal.acr.1001141
Copyright license: © 2025 Angel-Rodríguez F, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Synergistic Heterozygosity in Rare Digenic Hereditary Anemias Accurately Diagnosed by whole Exome Sequencing
Felipe Angel-Rodríguez1, Juliana Usta-Stavoli1, Andres Cruz-Gonzalez1, Gabriela Huguet2, Adriana Urbina2, Daniel Silgado-Guzman3, Dora Janeth Fonseca-Mendoza2, Nora Contreras-Bravo2, Luis Celis-Regalado1, Carlos M Restrepo2 and Adrien Morel2*
1School of Medicine and Health Sciences, University of La Sabana, Chía, Colombia
2Center for Research in Genetics and Genomics (CIGGUR), Institute of Translational Medicine (IMT), School of Medicine and Health Sciences, Rosario University, Bogotá, Colombia
3Department of Molecular Diagnosis, Molecular Genetics of Colombia SAS, Bogotá, Colombia
*Address for Correspondence: Adrien Morel, Ph.D, Professor, Center for Research in Genetics and Genomics (CIGGUR), Institute of Translational Medicine (IMT), School of Medicine and Health Sciences, Rosario University, Bogotá, Colombia, Email: [email protected]
Sickle Cell Disease (SCD) and its forms are the most common inherited blood disorders worldwide [1] and has a broad phenotypic variability among individuals regarding modifier genes and genetic mutations which change the severity spectrum [2]. Sickle Cell Trait (SCT) is found widespread in human populations including Colombia, which has received migrants from Europe, the Middle East, and Africa over the past five centuries, including recent decades [3].
Genetic tests for inherited anemias are performed on individual genes to confirm previous biochemical or electrophoretic findings [4]. However, two SCT cases came to our attention due to their symptoms, clinical severity, and frequency and intensity of acute crises, but the diagnosis was clarified only after Whole Exome Sequencing (WES) became available that could clarify the diagnosis and the variability in the phenotype explained.
Family A. A 37-year-old female with chronic abdominal pain since childhood, asthenia, and adynamia, with joint pain, peripheral edema, which limits daily activity. She worked in Bogota (altitude 2.620 meters a.s.l.) alternating with La Mesa (Cundinamarca) (1.200 asl), in higher altitude she noticed low growth in nails and hair, yellowish teeth color and xeroderma.
She had 3 pregnancies, from two different couples. During the first pregnancy, she experienced dyspnea and diaphoresis on exertion, intermittent blurred vision, macroscopic hematuria which persisted until 8 months after delivery, normocytic-normochromic anemia (hemoglobin level 7.5 gr/dl), which did not require transfusion support. The second was a dizygotic twin pregnancy, which presented preeclampsia, sudden blurred vision (with normal retinal angiography). Caesarean section was indicated at week 35. The female twin required neonatal reanimation due to sudden death and a sickle cell trait was identified in the neonatal screening followed by electrophoresis which showed presence of HbS in 44% (Figure 1A).
Family B. A 24-year-old male, who suffered anemia since he was born and received several blood transfusions in the first month and then at two months of life. He was recorded with low hemoglobin and hematocrit counts when he lived at sea level, and showed a mild improvement when he returned to Bogota. He presents an acute disease with hemolysis, jaundice, abdominal pain and hepatosplenomegaly. Hemoglobin count under 10 mg/dl was recorded about ten times in twenty years. At the age of 19, a cholecystectomy was performed after cholelithiasis. His father is 61-year-old, with ancestry from Perpignan (France) and his mother 53-year-old was identified as a carrier of the sickle cell trait. They were non-consanguineous and healthy as well as his sister with 21 years (Figure 1B). A maternal first cousin has sickle cell trait.
Both families received Whole Exome Sequencing (WES) followed by Sanger sequencing analysis for HBB, HBA, PKLR and SPTA1 genes. Once the mutations were identified in both families, classical laboratory tests were done to confirm the genetic findings, blood cell count, hematocrit, peripheral smear blood, hemolysis profile, MCV, and pyruvate kinase activity (Table 1).
Family A. Showed the patient (III, 2), grandmother (I, 2: who had experienced an ischemic attack), and dizygotic twins (IV, 2 and IV, 3) all were SCT carriers. III, 2 and IV, 2 also have PKLR mutation (Figure 1A). Index case paternal family was originated in Italy and were presumably healthy. II, 2 suffered central retinal vein occlusion.
Whole Exome Sequencing (WES) was performed in the index case (III, 2) revealed two mutations. First, the patient is a carrier of heterozygous pathogenic variant for the gene HBB: c.20A>T p.Glu7Val (SCT). Second, heterozygous pathogenic variant was identified for the gene PKLR: c.1456C>T p.Arg486Trp which concludes the diagnosis of an uncommon hemolytic anemia caused by a biallelic digenic mutation (Figure 1A). Both findings were confirmed by Sanger sequencing (Figure 1A). Then, dizygotic twins were sequenced, both showed as carriers of SCT (HBB: c.20A>T p.Glu7Val mutation) and the daughter have PKLR: c.1456C>T p.Arg486Trp as well as her mother, who was also affected (data not shown).
Family B. Index case (II, 1) WES and Sanger sequencing showed two heterozygous mutations: the common mutation HBB c.20A>T p.Glu6Val (SCT) and SPTA1: c.83G>A p.Arg28His (Figure 1B).
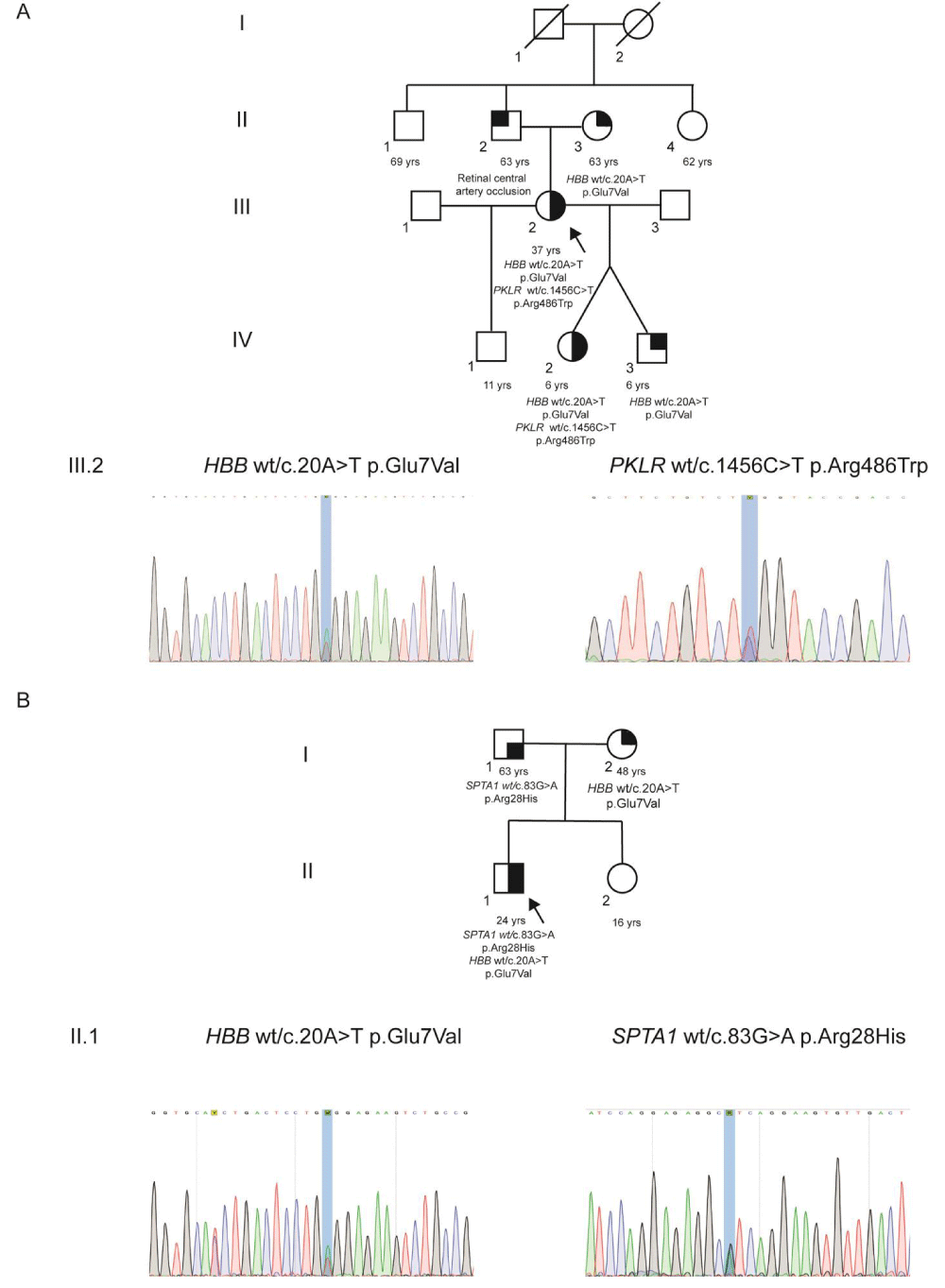
Figure 1: Familial segregation and Sanger sequencing results for HBB, PKLR, and SPTA1 variants A. Pedigree and mutation profile of Family A and Sanger sequencing of the index case III,2. B. Pedigree of the Family B and Sanger sequencing of the index case II,1.
Afterwards, blood laboratory analyses in Family A, III, 2 and IV, 2 showed Hemoglobin electrophoresis revealed HbS levels of 40% and 38.6%, respectively. PKLR activity was 8.7 and 10.1 U/g Hb (37 ºC) respectively (Table 1). Family B, II, 1 laboratory test data showed HbS 39.9% and increased levels of indirect, direct and total bilirubin. Other data such as HBA reduction and increased HbS levels are shown in Table 1.
| Table 1: Relevant data from laboratory tests. | ||||
| Analytics | Family A | Family B | Reference values | |
| Patient 1 | Patient 2 | Patient 3 | ||
| Blood count | ||||
| Hgb g/dl | 14.9 | 15.4 | 14.7 | 13.6 - 17.5 |
| PCV % | 43.2 | 44.5 | 42.2 | 41.0 - 53.0 |
| MCV fl | 91 | 80 | 80 | 80 - 98 |
| MCH pg | 31.4 | 27.8 | 28.9 | 24.0 - 30.0 |
| MCHC g/dl | 34.4 | 34.7 | 34.8 | 32.0 - 35.0 |
| WBC | 6.5 | 10.2 | 6 | 4.5 - 10.0 |
| Platelets | 320 | 420 | 332 | 150 - 450 |
| Haemoglobin electrophoresis | ||||
| HBA% | 56.5* | 57.5* | 56.9* | 96.8 – 97.8 |
| HbF% | 0.3 | 0.6 | 0.2 | Ad: < 2.0, Nb: 50.0-80.0, 6m: 0.0-8.0 |
| HbS% | 40* | 38.6* | 39.9* | - |
| HBA2% | 3.2 | 3.3 | 3 | 2.2-3.2 |
| Peripheral blood smear | ||||
| Erythrocyte abnormalities | Normal | Normal | Normal | |
| Bilirubin | ||||
| TB mg/dl | 0.83 | 0.82 | 5.12* | 0.2-1.1 |
| DB mg/dl | 0.27 | 0.3 | 1.04* | 0.0-0.3 |
| IDB mg/dl | 0.56 | 0.52 | 4.08* | 0.0-0.9 |
| Pyruvate kinase activity | ||||
| (U/g Hb at 37°C) | 8.7 | 10.1 | - | 7.4-16.4 |
| Hgb-Hemoglobin concentration, PCV: Packed Cell Volume; MCV: Mean Cell Volume, MCH: Mean Cell Hemoglobin; MCHC: Mean Cell Hemoglobin Concentration; WBC: White Blood Cells*10⁹/l, Plts: Platelets*10⁹/l. Ad: Adults; Nb: Newborn; 6m: 6 months. TB: Total Bilirubin; DB: Direct Bilirubin: IDB: Indirect Bilirubin. | ||||
Gene mutations that cause hereditary anemias are worldwide distributed. Colombian population is mixed by Amerindians and diverse migrations from Europe, Africa and Asia. In both families, A and B, were identified SCT carriers, but index cases and other relatives showed a clinical course of symptomatic critic severe anemia that worsened with time? Their severe symptoms were misunderstood, and it even took several decades to clarify clinical-laboratory incoherence, that suggested an additional hematologic disease. It was until WES was performed that causal mutations were detected using a multigenic panel for inherited anemias.
In Family A digenic disease was identified, two members presented SCT and PKLR deficiency simultaneously (HBB: c.20A>T p.Glu7Val and PKLR: c.1456C>T p.Arg486Trp). The male dizygotic twin, who was also studied, carried only sickle cell trait (HBB: c.20A>T p.Glu7Val). Those findings led to a diagnosis of pyruvate kinase deficiency, likely exacerbated by SCT. The great-grandfather of patient III, 2 had Italian origin. In Italy, SCT prevalence reaches peaks of 13%; however, recent international migration flows may change these data [5]. In Colombia, the SCT prevalence in an isolated afro-descendant population in the southern region, showed data up to 10% [3]. PKLR: c.1456C>T prevails in south Europe, which is consistent with the paternal ancestry of Family A [6]. PK deficiency is the most common glycolysis associated defect that causes non-spherocytic chronic hemolytic anemia, with a prevalence of 1:20.000 in the Caucasian population. Pyruvate kinase is involved in the second-to-last step of glycolysis, responsible for ATP production. Symptoms depend on hemolysis severity, which can range from asymptomatic to life-threatening fetal or neonatal anemia and has an autosomal recessive inheritance pattern [6,7].
Cases of this digenic biallelic anemia as presented here, are rare and primarily reported in patients of African or Mediterranean ancestry. Co-inheritance of SCT and PK deficiency has been previously reported in other reports previously [7]. In addition, a 29-year-old Moroccan woman presented frequent occlusive crises, bone pain, skin ulcers, splenomegaly, and moderate anemia (hemoglobin 9.0 g/dl). Hemoglobin electrophoresis showed 42% HbS and 58% HBA, and genetic analysis revealed the same digenic biallelic mutations in PKLR and HBB genes [8]. These cases support an interaction between two genetic defects in both red blood cell metabolism and hemoglobin disturbed pathophysiology. This may be related to high 2,3-diphosphoglycerate (2,3-DPG) levels due to PK deficiency which reduces oxygen affinity and precipitates HbS polymerization and sickling, leading to severe anemia despite SCT carrier status [7,9]. In addition, PK deficiency can lead to increased fragility and hemolysis of sickled RBCs [8]. Family A was undiagnosed for at least three decades a biallelic digenic condition was identified through WES.
Family B, a second case of digenic biallelic anemia which was undiagnosed for two decades, also showed SCT (HBB:c.20A>T p.Glu6Val) and SPTA1:c.83G>A p.Arg28His, resulting in a hereditary elliptocytosis due to spectrin dysfunction, worsened by SCT. Mother’s the index case’s mother was a carrier of SCT, and the father, who was asymptomatic, segregated the autosomal dominant SPTA1 gene mutation.
Gene encoding alpha-spectrin (SPTA1) results in a defective horizontal association of the RBC cytoskeleton, leading to membrane deformability, stability and increased hemolysis causing Hereditary Elliptocytosis (HE) and RBC cytoskeletal disorders [10]. Weakening of the spectrin-based cytoskeleton by the SPTA1 mutation would be expected to contribute to the aggravated hemolysis observed in co-inheritance with HBB SCT, a second example of digenic biallelic anemia, resulting in polymerization of mutant hemoglobin under hypoxic conditions and the damage to red blood cell cytoskeletal proteins [11].
The presence of two pathogenic variants in both genes HBB and PKLR for Family A, and HBB and SPTA1 for Family B, are examples of combination of two different alleles in a double heterozygous individual, resulting in a synergistic effect with implications for the severity or expression of a disease, known as synergistic heterozygosity [12].
Some of classic laboratory analysis for SCT/SCD are sickle solubility testing, hemoglobin electrophoresis, High-performance Liquid Chromatography (HPLC), and Isoelectric Focusing (IEF) may not be readily available in all clinical settings and health facilities. Currently, sickle solubility tests are limited in that since it cannot differentiate patients with SCT from SCD; and infants with high hemoglobin F and low hemoglobin S may result in false negatives, requiring confirmatory testing [13,14]. Molecular protocols for hemoglobinopathies are often used in the research setting (archived DNA samples) to identify SCT carriers and are also used by a limited number of laboratories to clarify SCT screening in rare anemia cases [15]. Recent genomic advances have enhanced the diagnosis of SCT, SCD, thalassemia and other rare anemias using WES [13], as happened in the present two families who received a precision diagnosis, reducing diagnostic delays and streamlining laboratory workflows. Molecular testing, particularly WES or anemia panels, is recommended, particularly WES and/or a multigenic anemia panel in all cases of symptomatic anemias with or without SCT.
Contributions
FA, JU, AC, AM, GH, AU, and CR contributed to manuscript writing. DF, DS, NC, AM, AU and LC participated in laboratory tests, molecular analysis and bioinformatics. FA, JU, AC, AM, GH, AU and CR participated in patients care. All authors reviewed and approved the final manuscript.
Data-sharing statement
Data will be shared upon reasonable request submitted to the corresponding author upon request.
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017;376(16):1561–73. Available from: https://doi.org/10.1056/nejmra1510865
- Onimoe G, Rotz S. Sickle cell disease: A primary care update. Cleve Clin J Med. 2020;87(1):19–27. Available from: https://doi.org/10.3949/ccjm.87a.18051
- Fong C, Lizarralde-Iragorri MA, Rojas-Gallardo D, Barreto G. Frequency and origin of haplotypes associated with the beta-globin gene cluster in individuals with trait and sickle cell anemia in the Atlantic and Pacific coastal regions of Colombia. Genet Mol Biol. 2013;36(4):494–7. Available from: https://doi.org/10.1590/s1415-47572013000400005
- Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4(1):18010. Available from: https://doi.org/10.1038/nrdp.2018.10
- Pinto VM, De Franceschi L, Gianesin B, Gigante A, Graziadei G, Lombardini L, et al. Management of the Sickle Cell Trait: An Opinion by Expert Panel Members. J Clin Med. 2023;12(10):3441. Available from: https://doi.org/10.3390/jcm12103441
- Zanella A, Bianchi P, Fermo E. Pyruvate kinase deficiency. Haematologica. 2007;92(6):721–3. Available from: https://doi.org/10.3324/haematol.11469
- Manco L, Vagace JM, Relvas L, Rebelo U, Bento C, Villegas A, et al. Chronic haemolytic anaemia because of pyruvate kinase (PK) deficiency in a child heterozygous for haemoglobin S and no clinical features of sickle cell disease. Eur J Haematol. 2010;84(1):89–90. Available from: https://doi.org/10.1111/j.1600-0609.2009.01353.x
- Alli N, Coetzee M, Louw V, van Rensburg B, Rossouw G, Thompson L, et al. Sickle cell disease in a carrier with pyruvate kinase deficiency. Hematology. 2008;13(6):369–72. Available from: https://doi.org/10.1179/102453308x343536
- Cohen-Solal M, Préhu C, Wajcman H, Poyart C, Bardakdjian-Michau J, Kister J, et al. A new sickle cell disease phenotype associating Hb S trait, severe pyruvate kinase deficiency (PK Conakry), and an alpha2 globin gene variant (Hb Conakry). Br J Haematol. 1998;103(4):950–6. Available from: https://doi.org/10.1046/j.1365-2141.1998.01094.x
- Risinger M, Christakopoulos GE, Schultz CL, McGann PT, Zhang W, Kalfa TA. Hereditary elliptocytosis-associated alpha-spectrin mutation p.L155dup as a modifier of sickle cell disease severity. Pediatr Blood Cancer. 2019;66(2):e27531. Available from: https://doi.org/10.1002/pbc.27531
- Ustun C, Kutlar F, Holley L, Seigler M, Burgess R, Kutlar A. Interaction of sickle cell trait with hereditary spherocytosis: splenic infarcts and sequestration. Acta Haematol. 2003;109(1):46–9. Available from: https://doi.org/10.1159/000067273
- Vockley J, Rinaldo P, Bennett MJ, Matern D, Vladutiu GD. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol Genet Metab. 2000;71(1–2):10–8. Available from: https://doi.org/10.1006/mgme.2000.3066
- Naik RP, Haywood C. Sickle cell trait diagnosis: clinical and social implications. Hematology Am Soc Hematol Educ Program. 2015;2015(1):160–7. Available from: https://doi.org/10.1182/asheducation-2015.1.160
- Tubman VN, Field JJ. Sickle solubility test to screen for sickle cell trait: what’s the harm? Hematology Am Soc Hematol Educ Program. 2015;2015:433–5. Available from: https://doi.org/10.1182/asheducation-2015.1.433
- Benson JM, Therrell BL. History and current status of newborn screening for hemoglobinopathies. Semin Perinatol. 2010;34(2):134–44. Available from: https://doi.org/10.1053/j.semperi.2009.12.006